







Arrhythmogenic cardiomyopathy (ACM) is usually referred to as arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D).1A first historical description was made in 1736, whereas its first modern description dates back to 1982.2 Initially, ACM was thought to be an embryological malformation.3 Yet in recent years it became evident that the pathophysiology of an ongoing genetically determined myocardial atrophy did not fit the theory of a congenital myocardial aplasia. Genetic and pathological studies have been crucial to understand ACM. From autoptic studies we know that atrophy of the ventricular myocardium due to progressive myocyte loss and infiltration by fibrofatty tissue are key findings.4 These studies led to the assignment of ARVC/D as a primary cardiomyopathy by the World Health Organization in 1995.5
In its most typical form, the right ventricle (RV) is primarily affected, which is then referred to as ARVC/D.6 As the disease progresses, the left ventricle (LV) may also be involved.7 Molecular studies have identified causative mutations in genes encoding proteins of the intercalated disc, desmosomes in particular.8 These mutations impair the electrical and mechanical stability of the ventricular myocardium9,10 with subsequent inflammation, apoptosis, necrosis and fibrofatty infiltration, which usually begins after puberty.11,12 In a minority of patients non-desmosomal mutations in calcium regulating genes, growth factors and other structural genes have been associated with ACM. Nonetheless, genetic mutations cannot entirely account for phenotypic expression and disease progression, and genetic mutations cannot be identified in up to 50 % of the ACM population studied. Therefore, epigenetic and environmental factors such as exercise seem to play a pivotal role as disease modifiers.1,13,14 Increased workload for the myocardium during physical activity enhances this adverse remodelling, and the thinner RV is particularly prone.15 Due to the multiple facets of the disease, the term ARVC/D is somewhat misleading. Since biventricular involvement and LV involvement may be present,1,16 a broader term such as arrhythmogenic cardiomyopathy has been recently proposed by the Heart Rhythm Society/European Heart Rhythm Association (HRS/EHRA).1,16,17 Together with novel genetic evidence that further expanded the pathophysiology of this heterogeneous disease beyond desmosomal mutations, the term ACM is now commonly being used to describe a hereditary form of non-hypertrophic cardiomyopathy that primarily manifests with ventricular arrhythmias due to fibrofatty infiltration of the ventricular myocardium.1,18,19
Epidemiology
Phenotypic expression is more common in males (2–3:1).14 ACM usually manifests during adolescence, but can also emerge in the elderly.20 With a general prevalence of 1:2,000–1:5,000 ARVC/D is a rare disease, which is defined as a prevalence of ≤1:2,000 according to the European definition.21 However, in some endemic areas the prevalence may be higher.22 ACM is a leading cause of sudden cardiac death (SCD) due to ventricular tachyarrhythmias, particularly in young athletes ≤35 years of age.23,24 In one Italian study, ACM accounted for up to 22 % of SCD in young adults.6,25,26 Generally, ACM first manifests with ventricular arrhythmias. In classical ARVC/D, ventricular arrhythmias arise from the RV.27
Genetics and Disease Modulation
ARVC/D is usually inherited as an autosomal dominant trait. Recessive forms with additional phenotypic characteristics exist.28 Recessive mutations of desmosomal genes can cause severe forms of ARVC/D, such as Naxos disease and Carvajal syndrome, some with skin and hair involvement, since both alleles are involved.29,30 Moreover, it has been shown that in up to 18 % of patients with ARVC/D, compound or digenic heterozygosity is present, indicating that in some cases more than one pathogenic allele may be involved.31 Currently 13 different genetic loci have been reported to be associated with ARVC/D. The most common genes encode for desmosomal proteins.32,33 Recent studies have shown that non-desmosomal genes can also lead to an ARVC/D phenotype (see Table 1). However, in cardiomyopathies – similar to other genetic diseases – it is very important to use rigorous criteria when calling a genetic variant or mutation pathogenic, since many variants may only constitute innocent bystanders.
A recent transatlantic collaboration with ACM registries from the Netherlands and the US has suggested rigorous criteria on how to classify genetic variants/mutations in ACM, which are now widely accepted in this field.32 Another recent large-scale trial on long QT and Brugada syndromes from the US has revealed that even among laboratories experienced in genetic testing for cardiac arrhythmia disorders, there was low concordance in designating variants as pathogenic. In an unselected population, the putatively pathogenic genetic variants were not associated with phenotypic abnormalities, which raises questions about the implications of notifying patients of incidental genetic findings.1,34,35 Thus, it is important to consider whether the genetic finding is present in a symptomatic patient or family member with a rather high pre-test probability, or in an asymptomatic patient as an incidental finding, who as a matter of course has a low pre-test probability.35
The American College of Medical Genetics and Genomics has published recommendations on how to report incidental findings in clinical exome and genome sequencing.36 Genetically affected relatives demonstrate variable, and often milder phenotypes, which has been attributed to incomplete disease penetrance. Therefore, the prevalence of familial disease is probably underestimated.32 Nonetheless, an important reason why index patients usually present with more severe phenotypes than their family members is so-called ascertainment bias. The clinician should bear in mind that relatives due to ascertainment alone will likely have milder phenotypes since they were not the ones to initially present. As opposed to this, index patients (probands) by default are always the most affected, which is an important reason why some genetics literature would exclude probands in analyses to offset ascertainment bias. This sort of bias is inherent to genetic diseases and has to be considered when performing genetic testing of index patients and family cascade screening.37
Desmosomes provide cell-to-cell adhesion and consist of three major groups of proteins:38 a) the transmembrane proteins (cadherins) desmocollin-2 (DSC2) and desmoglein-2 (DSG2), b) desmoplakin (DSP) and c) the linker armadillo proteins plakoglobin (JUP) and plakophillin-2 (PKP2), which are mediators between the cadherins and DSP.9,39 In about 80 % of cases with confirmed pathogenic mutations, mutations in PKP2, DSP, and DSG2 are identified,8 with the most common mutations involving PKP2. Several mutations have been described in these desmosomal genes. Besides unambiguous pathogenic splice site and truncating mutations, it is crucial to also identify pathogenic missense mutations in patients with ACM, as they have been reported to have the same impact on disease outcome as other mutations.32,40 Yet, the variability of genotype–phenotype correlations and the heterogeneity of genetic mutations pose a great challenge for ACM diagnosis and risk stratification. Some mutations have been associated with specific clinical outcomes. For instance, truncating mutations in DSP or digenic/ compound heterozygosity of desmosomal genes are associated with more aggressive phenotypes and can be considered as risk factors of SCD and heart failure.32,41,42 Further studies with larger patient cohorts and rigorous genetic criteria are needed in order to improve our understanding of genotype–phenotype correlations in ACM.
In a minority of cases, mutations can be detected in non-desmosomal genes. Mutations in the ryanodine receptor, which releases calcium from the sarcoplasmic reticulum during muscle contraction,43 and in the key calcium regulating protein phospholamban (PLN) may also lead to ACM phenotypes, and although these patients may present at an older age, their long-term prognosis might be worse.32,44 Mutations in transforming growth factor β 3 (TGFβ3) have also been reported, resulting in its overexpression inducing myocardial fibrosis by stimulation of mesenchymal proliferation.
TGFβ3 modulates desmosomal expression as well, so mutations can alter desmosomal distribution and influence cell–cell stability.45 A mutation in the transmembrane protein43 (TMEM43) leads to a severe and highly lethal form of ARVC/D. This mutation was identified as a founder mutation in Newfoundland. Although this mutation is not located on the heterosomes, the phenotype is gender-specific with males having lower life expectancy than females, and carrying a higher risk for SCD.46 Although the pathophysiological role of TMEM43 remains uncertain, studies have proposed that it may interact within the adipogenic pathway and also lead to nuclear structural changes.47 The TMEM43 p.S358L (Newfoundland) mutation has been shown to be fully penetrant.48 In this subgroup, it is important to screen large pedigrees since this mutation can be highly lethal and often presents with SCD as a first symptom.49 Newer data shows that further proteins are associated with ACM. The intracellular filament protein desmin, α-T catenin (a cytoplasmic protein), lamin A/C (a nuclear protein) and titin (a large sarcomeric protein) have been associated with ACM and phenocopies.1,34,50
Various genetic mutations have been associated with ACM, but these cannot account for the entire spectrum of disease expression. Therefore, epigenetic and environmental factors may act as disease modulators. First evidence for this hypothesis arose from monozygotic twin studies, where differences were reported in symptom onset, disease severity and arrhythmic risk.51 A male predominance of disease expression has been generally described in ACM, with male gender constituting an independent risk factor for adverse outcome.52,53 Recent data of compound and digenic heterozygosity indicates that modifier genes may account for residual variation and disease severity.31,54 Most importantly, strenuous physical activity, particularly endurance sports, plays an important role for early disease manifestation, disease severity and progression.13,55,56
Understanding the relationship between genotype and phenotype is challenging. Although ACM is mostly inherited as an autosomal dominant trait, this is likely an oversimplification since disease expressivity and penetrance are generally low. Clinical presentation and disease course can substantially differ within the same affected family due to the complex genetic and epigenetic background. Yet, it should be noted that variable phenotypic expression is seen in almost all genetic diseases, and is not only encountered in ACM. Using genetic testing as a diagnostic tool can be challenging. This particularly holds true for next-generation sequencing (NGS) methods, by which an abundancy of genetic variants of unknown significance can be detected, which is particularly challenging when it comes to the interpretation of borderline or overlapping phenotypes.34,57 Therefore, as previously mentioned, rigorous criteria to label a mutation as pathogenic should be used. Data from exome sequencing projects and in silico predictive programs can be helpful in this regard, especially for missense mutations and variants in order to avoid 'genetic' overdiagnosis.1,32 Nevertheless, genetic testing may be very helpful to verify ACM in the index patient and in identifying affected relatives and subclinical/ concealed phases. It is hoped that NGS will facilitate the identification of new disease modifiers or causative mutations, particularly in patients in whom a desmosomal mutation is present, but disease penetrance is not complete in family members harbouring this particular desmosomal mutation, and the real genetic culprit is yet to be found.58,59
Pathogenesis
The desmosomal complex is crucial for cellular adhesion, tissue strength and stability. It is located at the cardiac intercalated disc. A defective desmosome can result in a loss of cardiomyocytes accompanied by fibro-fatty tissue replacement.9,60,61 Desmosomal proteins interact with other junctional molecules such as other cadherins, catenins, ion channels, e.g. the sodium channel Nav1.5, and gap junction molecules.62 A functional link between desmosomes, gap junctions and Nav1.5 has been described, and recently named connexome.63 Several mechanisms are being discussed in the pathogenesis of ACM. Reduced cell-to-cell adhesion by desmosomal dysfunction due to genetic mutations is thought to be a key mechanism.39,64 Mutations in desmosomes change the 3D structure, their length and the total amount of desmosomes.65,66 This can trigger intercalated disc remodelling, which does not only alter mechanical stability, but also electrical coupling between cells, and intra/intercellular signal transduction affecting apoptosis and lipid metabolism.39,61,67,68
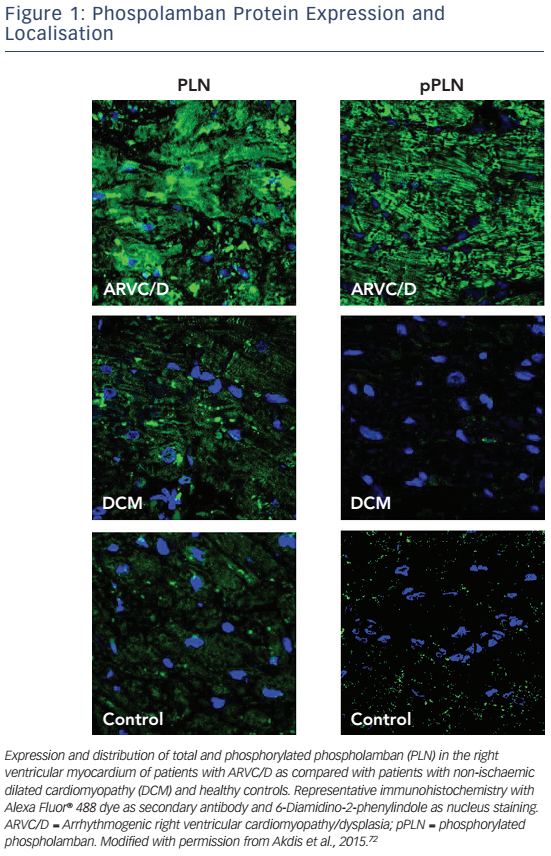
A decrease of JUP at the intercalated disc with its nuclear translocation and suppression of the canonical Wnt signalling pathway has been reported, even in individuals with other desmosomal mutations.65 Early histology and electron microscopy studies revealed that fibrofatty replacement of the RV goes along with inflammation and apoptosis.4,69,70 Induced pluripotent stem cell models from patients harbouring a pathogenic PKP2 mutation have highlighted that enhanced mitochondrial fatty acid uptake may lead to changes in fatty acid oxidation and promote fatty infiltration.61 Further experimental and clinical data suggest that non-desmosomal structures such as Nav1.5, PLN, desmin, titin and TMEM43 also interact with desmosomes and that mutations in these genes may change intracellular signalling pathways (see Figure 1).71,72PLN knockout has been associated with calcium channel modulation leading to VT.73 Furthermore, PLN phosphorylation has been reported to be upregulated in patients with ARVC/D72 in analogy to the pathophysiology of heart failure, which can be the endstage clinical presentation of ACM.74 Regarding titin mutations it has been shown that certain mutations may lead to a reduced structural stability and increased susceptibility for undergoing proteolysis.75
TMEM43 has also been associated with the intercalated disc and the adipogenic pathway.47,48 It has been shown that the TMEM43 p.S358L (Newfoundland) mutation may affect the localisation of proteins involved in cellular conduction and reduce conduction velocity in cardiac tissue.76 Yet, the role of non-desmosomal genetic mutations in the disease pathogenesis has not been fully elucidated, and there is a need for further experimental and clinical studies. Patient specific induced pluripotent stem cell models may play an important role in the discovery of novel disease mechanisms.61 Furthermore, volume overload and mechanical stress have also been suggested to enhance desmosomal dysfunction, which may explain why vigorous exercise has a negative impact on ACM phenotype, and why endurance athletes are particularly prone to adverse remodelling.77 In mice with PKP2 mutations, exercise leads to RV dilation and dysfunction.78,79 Clinical studies underscore the negative impact of sports, particularly high-intensity endurance training on ACM phenotype. 13,80 Myocarditis has also been associated with ACM, especially in genotype elusive patients, since cardiotropic viruses and bacteria have been found in cardiac tissue of these patients.11,81 Yet, the role of cardiotropic agents in the pathogenesis of ACM needs further investigation.
Phenotypic Expression
Classification of ACM into three different subtypes has been suggested. Right-dominant ARVC/D is considered as the classical form. Nonclassical forms were recently described.38,82,83 LV involvement is reported with a prevalence of up to 70 % of cases,84 which may be attributed to improved diagnostic methods.85 Yet, the proposed classification below is simplistic. It is important to keep in mind that due to genetic heterogeneity, epigenetic and environmental modifying factors and ascertainment bias there is a phenotypic continuum with the right- and left-dominant subtypes at opposite ends. Some mutations have been described to confer all described phenotypes.46 With regard to the important issue of ascertainment bias, individuals seeking medical attention often present with symptoms and advanced phenotypes. This can cause a selection bias in registries, and studies investigating ACM and genotype–phenotype correlations derived from these studies may not apply to a general ACM population. Such a bias is difficult to avoid in rare diseases such as ACM but has to be considered when interpreting study results.37
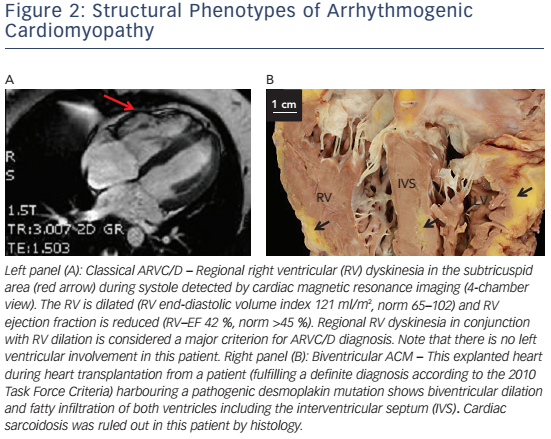
In right-dominant ARVC/D, a dilated RV with regional wall motion abnormalities with no or minimal LV involvement is observed (see Figure 2A). Myocardial remodelling starts in the subepicardial layers and may become transmural later on.17 Myocardial wall thinning can be seen on macroscopic examination.22,86 The subtricuspid region and RVOT are particularly prone to this remodelling process, leading to aneurysm formation.17 The concept of RV apical involvement and the term 'triangle of dysplasia' have recently been questioned.87 If an affected region is accessible for histological evaluation, inflammation, fibrosis and/or fatty infiltration can be visible.18,88
Biventricular arrhythmogenic cardiomyopathy is characterised by early involvement of both ventricles (see Figure 2B).89,90 Disease progression is characterised by systolic impairment and biventricular dilation with clinical features of global congestive heart failure. In contrast to dilated cardiomyopathy (DCM) with biventricular involvement, ventricular arrhythmias of both, right bundle branch block (RBBB) configuration – originating in the LV – and left bundle branch block (LBBB) configuration are present at an early stage.
Left-dominant arrhythmogenic cardiomyopathy (ALVC) has been proposed as a distinct form of ACM. It is characterised by the early occurrence of LV involvement (arrhythmias precede gross structural alterations), when global RV function is preserved. Electrocardiographic (ECG) and structural findings are left-sided analogues to those observed in ARVC/D (see Table 2).89
Clinical Presentation
ACM should be suspected if the following symptoms occur, particularly in young athletes:
- Palpitations
- Arrhythmic (pre)syncope
- Aborted SCD
- Chest pain ± rise in cardiac biomarkers
- Presumed DCM with early onset and frequent ventricular arrhythmias
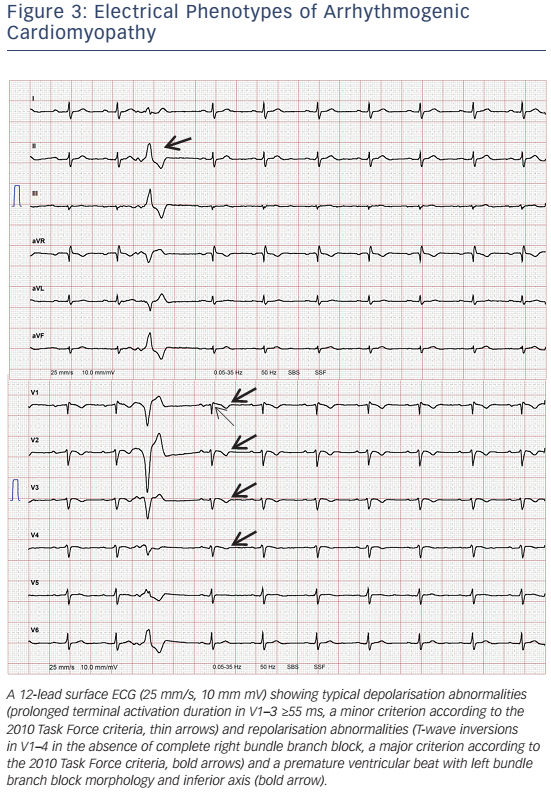
- Precordial T-wave inversions beyond V1 after puberty (see Table 2, Figure 3).
Although palpitations and (pre)syncope are the most frequent symptoms,91 they also occur in many other arrhythmic syndromes such as cardiac sarcoidosis, channelopathies and other cardiomyopathies.92 Since there is a considerable overlap between ARVC/D and 'arrhythmic forms' of idiopathic DCM, the broader term ACM has been introduced.18 It is important to distinguish syndromes leading to ventricular arrhythmias and SCD from benign forms and those forms primarily leading to heart failure.26,42,93 A high clinical suspicion should be raised if symptoms correlate with premature ventricular beats or VT, particularly LBBB morphology with a superior axis. However, left ventricular forms or biventricular disease can also present with VT with RBBB morphology. Monomorphic VT is associated with more advanced disease stages, although gross structural abnormalities are not always required for maintaining re-entry circuits.93,94 Up to onequarter of patients present with atrial arrhythmias, most frequently atrial fibrillation.95,96 This is associated with inappropriate implantable cardioverter defibrillator (ICD) shocks and an increased risk of both death and heart failure.97 It is not exceptional for ACM to manifest with SCD (annual incidence up to 9 %),98 both during strenuous physical activity99 and in the sedentary state.15 In ARVC/D caused by TMEM43 mutations, enhanced sympathetic activity has been shown as a trigger for lethal arrhythmias, particularly in males.47
T-wave inversions in V1–3 are benign until puberty, and their prevalence among athletes and controls seems to be similar thereafter.100 If T-wave inversions in V1–3 are detected after puberty, transthoracic echocardiography (TTE) can be performed to rule out structural heart disease. Dyspnoea and signs of right-sided heart failure are rare. Congestive heart failure may occur with progressive LV involvement. The treating physician should keep in mind that ACM cannot be excluded by the absence of structural abnormalities, as arrhythmias often occur in the early – so called 'concealed phase' – preceding structural abnormalities. In a study evaluating 37 ARVC/D families, <50 % of family members had overt disease and 17 family members did not display the phenotype despite harbouring the pathogenic mutation.101 Of note, during the concealed phase and in subtypes primarily presenting with heart failure, palpitations and (pre)syncope do not have a great diagnostic value. Therefore, it is important to combine several tools for diagnosis and risk stratification, including genetics whenever possible and reasonable. In certain subgroups a positive mutation status, particularly in males, may play a role in diagnosis and risk stratification,35,58,102 for example in families harbouring the TMEM43 p.S358L (Newfoundland) mutation – a fully penetrant, sex-influenced, highly lethal form of ARVC/D.48

Diagnosis
Revised 2010 Task Force Criteria
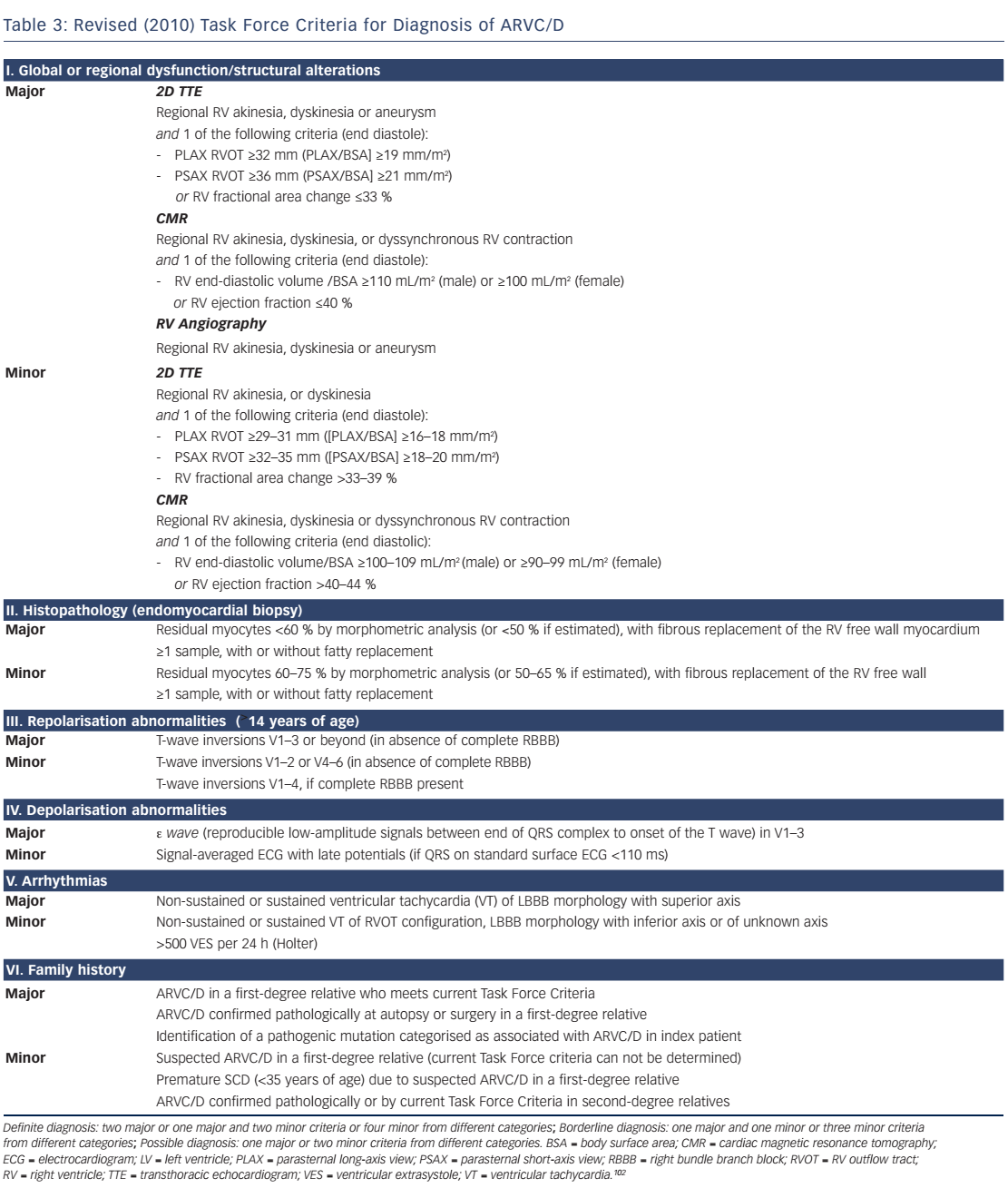
In 2010, the original 1994 TFC by McKenna et al.103 were revised to increase diagnostic sensitivity, particularly in affected asymptomatic family members.102 Pathogenic mutations were included, and cut-off values for imaging and histology were provided. The impact of these changes has been recently evaluated. Some investigators reported an increased diagnostic yield,104 whereas others did not.105,106 The clinician should bear in mind that the 2010 TFC only apply to ARVC/D, but not left-dominant ACM. For most forms of ACM there is only weak evidence supporting pre-symptomatic diagnosis by genetic testing.1,32 However, in the case of the TMEM43 p.S358L mutation, presymptomatic diagnosis can be made by a positive genotype only.48,49 For most subforms of ACM we have to rely on the 2010 TFC to establish the diagnosis. The current gold standard for diagnosis of ARVC/D are the 2010 TFC, at least until these criteria can be be further improved.102 The revised TFC consist of six diagnostic categories (see Table 3):
- Global and/or regional myocardial dysfunction and structural abnormalities
- Histological characterisation
- Repolarisation abnormalities on 12-lead surface ECG
- Depolarisation abnormalities on 12-lead surface ECG
- Arrhythmias
- Family history and genetics.
A definite diagnosis can be made with two major criteria, one major and two minor criteria, or four minor criteria from different categories; 'borderline' diagnosis with one major and one minor criterion, or three minor criteria and a 'possible' diagnosis if one major criterion or two minor criteria are present. Comprehensive non-invasive evaluation is mandatory, which includes a thorough clinical history, pedigree analysis, 12-lead surface ECG, TTE with detailed assessment of the RV, CMR, stress testing and Holter ECG. Event recorders and invasive diagnostics may be necessary if suspicion remains high and symptoms are rare. The revised TFC define quantitative criteria and abnormalities mainly based on comparison between adult index patients with ARVC/D and healthy controls. This approach has some limitations that should be taken into account. First, reference values of patients with ARVC/D and healthy subjects were derived from selected populations from tertiary care centres specialised in applying this particular diagnostic modality. Therefore, these reference values are prone to a substantial selection and ascertainment bias, and thus may not apply to the general ACM population, family members and individuals younger than 12 years. Moreover, they do not apply to patients with left-dominant forms.102
12-lead Surface ECG and Signal-averaged ECG
12-lead surface ECG will be abnormal in about 50 % of patients. This will include T-wave inversions in the right precordial leads, sometimes also involving V4–6, slurred S-wave upstroke in V1–3 ≥55 ms (see Figure 3), and with more advanced stages ε waves.107 The interpretation of ε waves significantly varies among observers. Furthermore, ε waves occur at more advanced stages, when the vast majority of patients already fulfils other TFC for definite diagnosis. Thus, ε waves in the absence of other diagnostic criteria should be interpreted with caution.108 T-wave inversions can be found in healthy individuals, patients with anterior ischaemia or RV hypertrophy.108,109 A recent study highlighted the importance of serial ECG evaluations, since dynamic ECG changes may occur.110 Delay of ventricular depolarisation due to scar (zig-zag pathways) may be visible as QRS fragmentation,108 ε waves111 or late potentials recorded by signal-averaged ECG (SAECG).112 SAECG may not be sensitive enough to detect early forms of ACM.112 Over recent years, the role of SAECG for ARVC/D diagnosis has diminished due to major advances in imaging and genetics.
Stress Testing
Ventricular arrhythmias in patients with ACM are often triggered by sympathetic activation. Thus, treadmill testing can reveal VT/VF or increase premature ventricular contractions with different morphologies. In a recent study, exercise testing revealed typical ECG abnormalities in a considerable number of patients with latent ARVC/D.113 In another study, the evaluation of the arrhythmic potential during very high dose isoproterenol infusion was sensitive for diagnosis of early forms of ARVC/D.114
Transthoracic Echocardiography
TTE is readily available in most centres and rapidly informative. Thus, TTE is considered as the initial imaging tool for suspected ACM and for screening family members. It may show RV enlargement and regional contraction abnormalities, most commonly in the subtricuspid region and RVOT.115 RVOT dimensions are crucial for diagnosis according to the 2010 TFC.116 The LV can be affected in up to 70 % of patients with ARVC/D displaying hypokinesia and a reduced ejection fraction. Frequently, LV structural abnormalities are localised in the posterolateral region.87,117,118 Novel technologies such as strain imaging allow for better quantification of regional wall motion, thereby allowing earlier disease detection and LV involvement.84
Cardiac Magnetic Resonance Tomography
Cardiac magnetic resonance tomography (CMR) has emerged as the non-invasive gold standard for assessing the RV over the past 20 years.119 Assessment of right-sided volumes and ejection fraction is highly accurate. Late-gadolinium enhanced (LGE) CMR can reveal myocardial fibrosis.120 Yet, myocardial fibrosis and fat as potential diagnostic features were not integrated in the TFC, because of their limited specificity, high intra-and inter-observer variability and the need for highly specialised interpreters.121,122 This owes to the fact that the RV is very thin and epicardial fat cannot reliably be distinguished from intramyocardial fat. However, CMR plays an important role for ARVC/D diagnosis (see Figure 2A). Consensus documents for nonclassic forms are awaited. The advent of novel technologies such as CMR tagging may facilitate early diagnosis of ARVC.123–127
RV Angiography
RV angiography is considered very useful to diagnose ARVC/D128 and thus is equivalent to TTE and CMR in the 2010 TFC. It has a positive predictive value of ∼85 %, with a negative predictive value of 95 %.129 High quality images allow assessment of RV morphology and wall motion. Yet, clinicians want to apply non-invasive diagnostic strategies without ionising radiation, particularly in young patients. Serial followup angiographies for monitoring disease progression are not feasible. Of note, hypokinesia is not considered diagnostic in the 2010 TFC, since akinesia or dyskinesia are required.
Electrophysiological Study and Electroanatomical Voltage Mapping
The goal of an electrophysiological study (EPS) in patients with ACM is the induction of sustained ventricular arrhythmias for making the diagnosis, risk stratification and to guide ablation. Moreover, the susceptibility for arrhythmias ± sympathetic challenge with isoproterenol, ICD treatment algorithms and efficacy of antiarrhythmic drugs (AAD) can be assessed. Electroanatomical voltage mapping (EAM) is a technique using electrophysiological catheters to measure myocardial voltages. After obtaining several hundred to thousands of points, a voltage map can be reconstructed. Normal myocardium generally displays bipolar voltages >1.5 mV. 130,131 In diseased myocardium, lower voltages with a longer duration, splitting and fractionation of signals can be recorded. Myocardial voltage maps can be obtained both from the endocardium and epicardium. EAM is generally safe, and improves outcomes of VT ablation.132–135 The diagnostic and prognostic utility of EAM has not been implemented in the current TFC, but recent data indicate that it can be useful for diagnosis and risk stratification.136
Endomyocardial Biopsy
Endomyocardial biopsy (EMB) has, for a long time, been considered the diagnostic gold standard for ACM diagnosis. Indeed, histological examination and immunostaining may allow confirmation of ACM, and exclude differential diagnoses, e.g. sarcoidosis, Chagas disease or other forms of myocarditis. Yet, biopsies are usually taken from the RV septum for safety reasons. Since the process of fibro-fatty infiltration spares the septum, EMB often yields false-negative results.137,138 EMB from diseased regions is problematic, as these regions are very thin, and sampling carries an increased risk of perforation. As ACM is patchy, several biopsies should be obtained. EAM-guided biopsies taken from low-voltage areas may improve diagnostic yield and better distinguish between myocarditis or sarcoidosis.130,139
Genetic Testing
When performing genetic testing, rigorous criteria to ascertain whether a mutation or variant is pathogenic should be used. An HRS/EHRA consensus statement for genetic testing in ACM was published recently.1 Genetic testing is performed to confirm ACM in individuals with a high (Class IIa) or intermediate (at least one major or two minor criteria; Class IIb) clinical suspicion and to identify genetically-affected relatives harbouring the pathogenic mutation (Class I). Genetic testing in cases fulfilling only one minor criterion is not recommended.8,42,58 A negative genetic test does not exclude ACM, since other unknown causal mutations and environmental factors may also cause the disease.60,140 With the exception of TMEM43 p.S358L mutation carriers48 and autosomal-recessive forms, genotype does not provide a diagnosis of ACM by itself.1,8,102 Yet, the identification of pathogenic mutations may be useful in the differential diagnosis of ACM and phenocopies, such as myocarditis, idiopathic RVOT tachycardia, DCM, muscular dystrophies or sarcoidosis.1,32,141 Genetic testing should not be seen as the only diagnostic tool, but may be very helpful especially in identifying affected relatives and subclinical/ concealed phases.58,59 Genetic cascade screening of relatives offers an alternative strategy to serial clinical evaluation. In this regard, the absence of a clear pathogenic mutation in a family member causing the disease in the index patient obviates the need for serial clinical evaluation in this family member. Current guidelines102 do not recommend genetic testing for risk stratification and therapeutic decision-making in ACM due to conflicting study results.8,142–144
Recent studies showed an association between positive genotype and earlier disease onset in index patients harbouring a known genetic mutation as compared with index patients without a known genetic mutation. However, ascertainment bias has to be considered in such a comparison.32 Moreover, these results can be biased by the fact that ACM is almost never fully penetrant, >40 % of ACM patients are genotype elusive, and the pathogenic mechanism in these patients is not clear. Yet, the results of genotype–phenotype correlation studies can be clinically helpful for some genetic mutations. For instance, truncating mutations in DSP, digenic or compound heterozygous mutations and mutations in the non-desmosomal gene TMEM43 are associated with more severe phenotypes and therefore, may be considered as risk factors of SCD.8,32,41,42,102 For TMEM43 p.S358L carriers, a diagnosis of ARVC/D can be made only by the presence of this genetic mutation, and clinical screening of family members is strongly recommended.49
Differential Diagnosis
A common differential diagnosis is idiopathic right ventricular outflow tract VT (RVOT-VT), particularly in early stages of ARVC/D lacking gross structural abnormalities. 145 RVOT-VT is not associated with structural heart disease, and thus has a more benign course. In RVOT-VT, 12-lead surface ECG and SAECG are normal during sinus rhythm. A single VT morphology with LBBB pattern and an inferior axis is recorded in most of the cases, although slightly different morphologies (all with an inferior axis) can occur if VT origin is above the pulmonary cusps.146 ECG scoring systems to differentiate idiopathic RVOT-VT and ARVC/D have been suggested.147 In ARVC/D, QRS duration during VT was longer (≥120 ms in lead I).148 Notching of the QRS and precordial transition in lead V6 were exclusively seen in ARVC/D. Idiopathic RVOT-VT is difficult to induce by programmed ventricular stimulation.148,149 Idiopathic RVOTVT responds well to verapamil. Endocardial ablation is feasible and curative, whereas in ARVC/D, ablation is not curative and an epicardial approach may be necessary to eliminate the clinical VT. Genetic testing, a positive family history, EAM and EMB can help to differentiate ACM and localised forms of myocarditis.150 Cardiac sarcoid can mimic ARVC/D, and the current TFC do not reliably distinguish between these two. In a study of patients with suspected ARVC/D also being evaluated by EMB, a high incidence (15 %) of cardiac sarcoid was identified.151 Cardiac sarcoid should be considered if respiratory and systemic symptoms, high-grade atrioventricular conduction block and septal involvement are present, and familial disease is absent. Diagnosis is confirmed by EMB and thoracic CT scans.151
DCM is particularly difficult to distinguish from non-classic forms of ACM, and these two entities can significantly overlap and harbour similar mutations. Palpitations, (pre)syncope and ventricular arrhythmias are present at an early stage in ACM, often in the absence of gross structural abnormalities, which is the opposite in DCM.18 Atrioventricular conduction block is more common in DCM, but mutations in lamin A/C can lead to ACM with conduction defects, as well.152 Brugada syndrome can mimic ACM, as RV conduction delay has been demonstrated in both entities. The presence of structural abnormalities favours ACM. Recently, a genetic overlap between these two has been suggested. In vitro studies have shown a cellular interaction between desmosomes and ion channels. Mutations associated with Brugada syndrome were found in ACM patients and vice versa.153 Titin mutations have also been associated with a phenotypic overlap between ACM and DCM. The same holds true for PLN mutations.44 However, the genetic interpretation of the large titin molecule and its variants is particularly challenging.75 Treatment of these patients and their arrhythmogenic risk has not been established yet, which has to be addressed in future studies. Other common differential diagnoses include right ventricular infarction, congenital left-to-right shunts, Chagas disease and Uhl’s disease.22 Finally, adaptation of the RV to increased workload in endurance athletes can mimic ARVC/D, and there is a debatable grey zone of what is considered physiological adaptation.154
Disease Course
ACM is a progressive disease however individual disease course can vary. Cardiac mortality is currently estimated ∼0.9 % per year. Most patients die of progressive heart failure or ventricular tachyarrhythmias.155 Four disease phases have been proposed:65
- Concealed phase: patients are asymptomatic and structural abnormalities are absent. SCD due to VF can be the primary manifestation in this phase.
- Occurrence of symptomatic arrhythmias
- Early heart failure symptoms
- End-stage heart failure.
Diagnosis and risk stratification during the concealed phase can be very challenging. Genetic testing with rigorous criteria and pre-participation screening of young athletes might help to identify affected individuals and prevent SCD.1,26,156 In one study, 7 % of ACM patients received cardiac transplantation after a mean follow-up of 10 years, mostly due to severe LV involvement.7 Physical activity promotes earlier disease manifestation and more rapid disease progression. Identification of affected athletes by pre-participation screening seems to substantially reduce mortality in this cohort.143,156
Therapy
Recently, an international consensus statement on the treatment of ARVC/D has been published.144 Data on non-classic ACM subtypes is scarce.
Physical Activity Restriction
Competitive athletes with ACM have up-to five-fold increased risk of SCD compared with sedentary individuals with ACM. Other studies have confirmed that high-level physical activity, particularly endurance sports, promotes disease onset, progression and adverse outcome.13,55 Thus, it is recommended that patients with a definite diagnosis of ARVC/D do not participate in competitive and/or endurance sports (Class I). Furthermore, they should be restricted from participation in athletic activities, with the possible exception of recreational low intensity sports (Class IIa).56,157
Pharmacological Therapy
Pharmacological options consist of antiarrhythmic drugs (AAD), e.g. sotalol, amiodarone and mexiletine, b-blockers and heart failure drugs. AADs are recommended as an adjunct to ICD therapy in patients with frequent discharges (class I).158 Moreover, they should be considered to improve symptoms due to frequent premature ventricular beats or non-sustained VT (class IIa), and may be considered as an adjunct to catheter ablation in selected patients with haemodynamically stable VT without an ICD (class IIb). Of note, cardioselective β-blockers should be considered in all patients with the ARVC/D phenotype irrespective of arrhythmias (class IIa), but their prophylactic use in healthy gene carriers is not recommended.158
Implantable Cardioverter Defibrillator
According to the most recent guidelines by the American College of Cardiology (ACC)/American Heart Association (AHA), the European Society of Cardiology (ESC) and the most recent international task force consensus statement on the treatment of ARVC/D, an ICD is recommended in patients with aborted SCD, haemodynamically unstable VT or VF (class I), and according to the consensus statement in those with severe systolic dysfunction of the RV, LV or both, irrespective of arrhythmias (class I).144,159–161 An ICD is generally indicated in ARVC/D patients who have experienced an episode of haemodynamically stable, sustained VT. If major risk factors such as unexplained syncope, moderate ventricular dysfunction, or non-sustained VTs are present, ICD implantation should be considered. Nonetheless, there is no clear consensus regarding primary prevention of SCD in ACM patients without documented sustained ventricular arrhythmia (VA).
The AHA recommends an ICD in patients with a familial cardiomyopathy associated with SCD (class IIb, level of evidence C).161 The ESC states that in patients without documented VA or syncope, an ICD may be considered after detailed clinical assessment including family history, severity of RV and LV function and other factors such as psychological health and socioeconomic status.160 According to the international consensus statement, ICD implantation may be considered in patients with minor risk factors such as gender, age and ECG abnormalities and these patients should be evaluated on an individual basis. A single-chamber device is generally preferred in order to minimise the incidence of lead-related complications in this relatively young cohort. Since many patients benefit from antitachycardia pacing, there is currently no role for subcutaneous ICDs. Importantly, prophylactic ICD implantation is not recommended in asymptomatic patients without risk factors and most healthy gene carriers (class III).144 However, since the TMEM43 p.S358L mutation is known to be fully penetrant and highly lethal, an ICD for primary prevention is indicated in all patients and family members harbouring this mutation (males immediately post-puberty and females ≥30 years) in order to improve survival in this subpopulation.49
Catheter Ablation
Catheter ablation of VT is recommended in patients with incessant VT or frequent appropriate ICD interventions for VT despite AAD (on amiodarone: class I, off amiodarone: class IIa).144 If ≥1 endocardial ablation procedure fails, an epicardial approach is recommended (class I).135 ARVC/D begins in the subepicardial layers. Thus, in experienced hands, a combined endocardial/epicardial approach can be followed as an initial ablation strategy (class IIa). Since catheter ablation is not curative, it is not recommended as an alternative to ICD for prevention of SCD.144
Conclusion
Discoveries within the last decade have substantially improved our understanding of ACM from a purely RV dysplasia to an inherited polygenic disease with a broad phenotypic spectrum. Although ACM predominantly affects the RV, atypical forms also affecting the LV are encountered. Emerging technologies in imaging, genetics and device therapy have facilitated diagnosis and prevention of SCD. Future challenges comprise early identification of asymptomatic patients and family members, improved risk stratification, and causal therapies to cure this challenging disease.
Clinical Perspective
- Arrhythmogenic cardiomyopathy (ACM) is a hereditary cardiomyopathy characterised by ventricular arrhythmias and structural/functional abnormalities of the ventricles.
- The most common arrhythmogenic right ventricular cardiomyopathy is generally referred to as ARVC/D.
- Causative mutations are detected in genes encoding the intercalated disc.
- Non-desmosomal mutations (e.g. TMEM43) have also been associated with ARVC/D and can cause malignant phenotypes.
- Exercise can promote disease onset, progression and adverse outcome.
- ACM is a common cause of sudden cardiac death in young athletes.
- The current gold standard for diagnosis are the 2010 TFC.
- Therapeutic strategies include restriction from endurance and competitive sports, β-blockers, antiarrhythmic drugs, heart failure medication, implantable cardioverterdefibrillators and endocardial/epicardial catheter ablation.