










AF is the most common sustained cardiac arrhythmia, affecting millions of people globally.1 It is associated with an increased risk of various adverse health outcomes, such as heart failure, stroke, dementia and other conditions, placing a substantial burden on healthcare systems worldwide.2
Despite significant advances in the development of drugs and interventional procedures for the treatment of AF, the therapeutic effects of these treatments remain suboptimal. The current management and prevention strategies for AF through upstream treatments highlight the importance of addressing risk factors. However, there is still much to be developed in terms of effective prevention strategies for AF.3
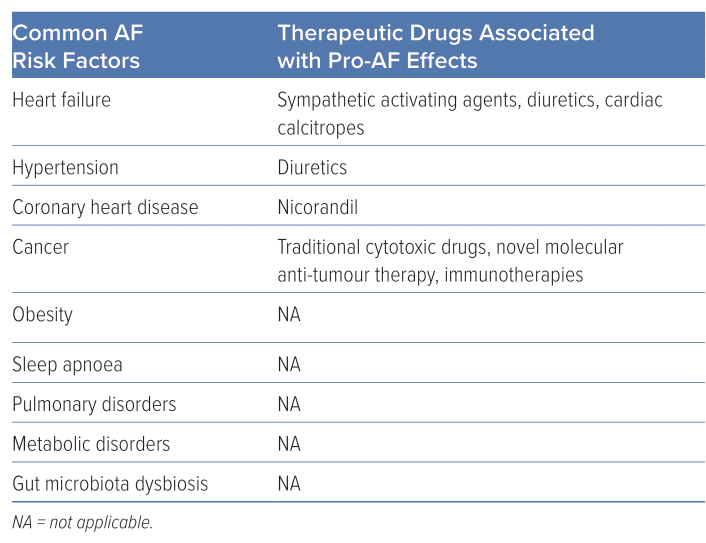
Risk factors for AF include advanced age, alcohol consumption, family history of AF, hypertension, thyroid dysfunction, sleep apnoea, structural heart diseases and gut microbiota dysbiosis (Table 1).4,5 Notably, drugs represent a significant risk factor for AF not mentioned in the latest AF guidelines.6 Drug-induced AF (DIAF) is a type of AF that is induced or precipitated by drug therapy and determining the underlying cause can be challenging. Despite the difficulty in diagnosis, DIAF can cause significant symptoms, which can have an impact on the management of relevant diseases. However, compared with other types of AF, DIAF is often more preventable and manageable once the causal drug is discontinued.
Drugs that might induce AF have been collectively reviewed in previous literature.7–10 The aim of this review is to comprehensively discuss DIAF, including potential molecular and electrophysiological mechanisms and suggested management strategies. By enhancing the understanding of DIAF, this review aims to raise awareness among healthcare providers of its possible occurrence and highlight the need for prompt and appropriate management.
Epidemiology
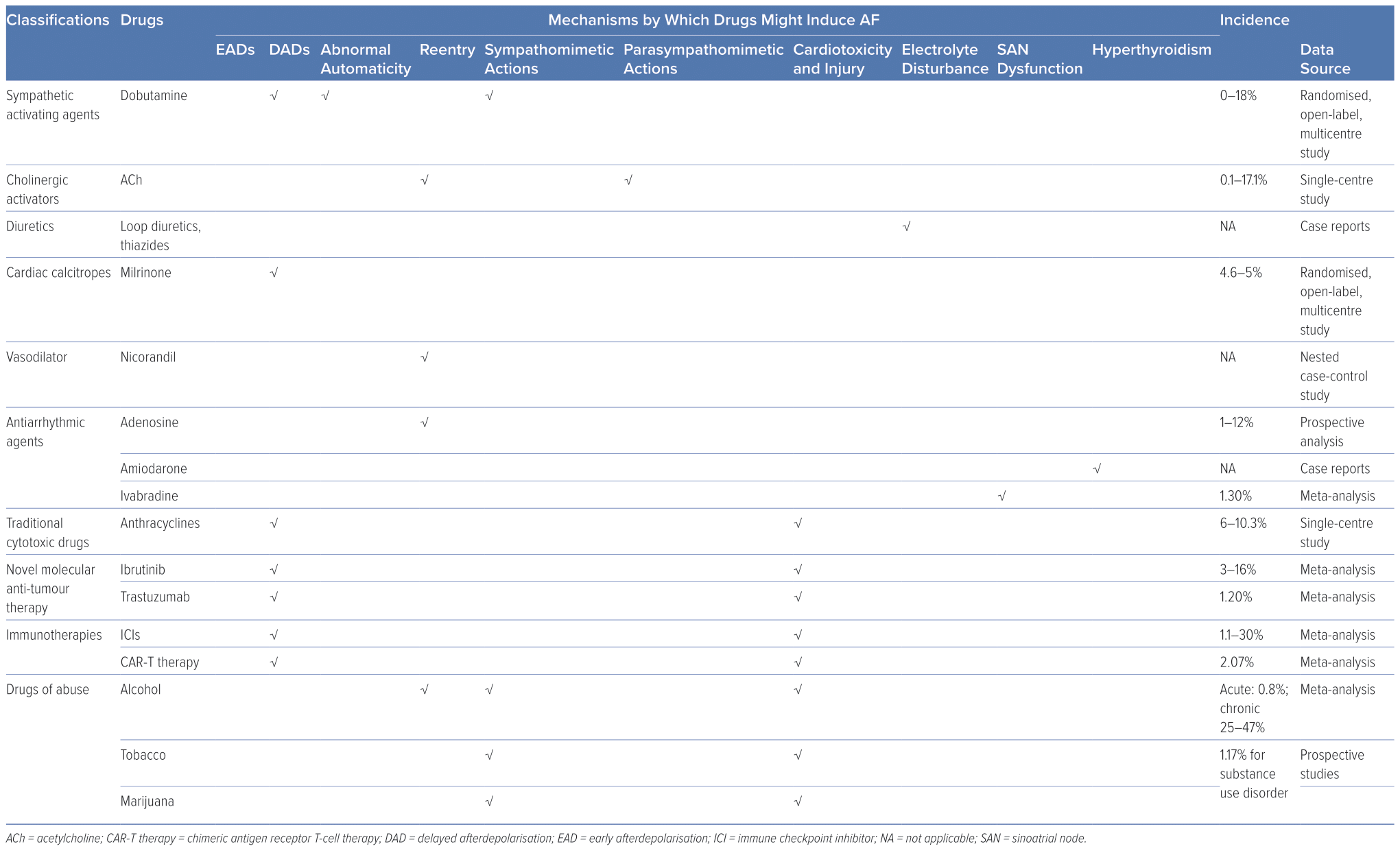
Assessing the prevalence of DIAF precisely and comprehensively is a formidable undertaking, as the available data pertaining to it are relatively scant. The incidence of DIAF is estimated to be relatively low and variable among different drugs. For example, the incidence has been reported as 0–18% for dobutamine, 4.6–5% for milrinone, 6–10.3% for anthracycline agents, 3–16% for ibrutinib, 1–12% for adenosine, 1.2% for trastuzumab, 1.3% for ivabradine, 0.1–17.1% for intracoronary acetylcholine (ACh), 1.1–30% for immune checkpoint inhibitors (ICIs), 2.07% for chimeric antigen receptor T-cell (CAR-T) therapy, 0.8% for acute alcohol consumption, 25–47% for persistent and heavy alcohol consumption and 1.17% for substance use disorder.10–29
It is worth noting that DIAF may exhibit a higher frequency among patients with coexisting severe cardiovascular disorders. For instance, in patients diagnosed with anthracycline-associated left ventricular dysfunction, the likelihood of developing AF has been reported to reach as high as 56.6%.30
Numerous accounts of DIAF rely on isolated case reports and substantial, all-encompassing clinical investigations are currently lacking. Consequently, the estimates of DIAF described above may not be entirely precise and are prone to underestimation. Additionally, the exact incidence of DIAF caused by other drugs is difficult to assess. Reasons for this may include – but are not limited to – the following:
- The prescription of many medications by non-cardiologists who may not be fully aware of the possible adverse effect of DIAF, leading to under-recognition of some cases.
- The symptomless nature of AF, which can be paroxysmal and asymptomatic (i.e., silent AF), unlike drug-induced QT prolongation, which may result in fatal ventricular arrhythmias with severe symptoms such as torsade de pointes; additionally, the incidence of AF is directly proportional to the frequency of screening, making it challenging to precisely evaluate its actual prevalence.
- Establishing a causal relationship between drugs and AF can be complicated because of the intricacies of pathophysiology, such as in patients with polypharmacy or in those with both diseases and relevant therapies that are associated with AF (for example, both cancer and anticancer treatment may induce AF), and because of the unavailability of a comprehensive medical history.31
As a result, despite the estimated low incidence of DIAF, pinpointing the precise prevalence remains a challenging task.
Mechanisms of AF and Drug-induced AF
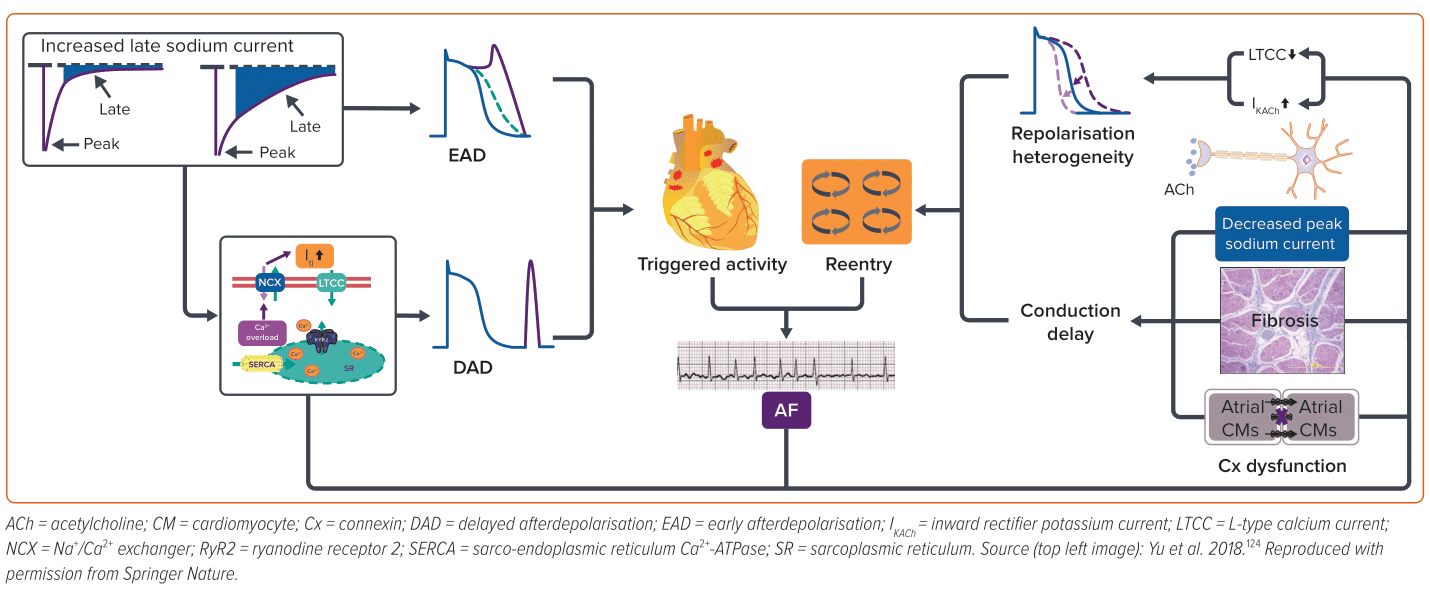
AF pathogenesis is complex and multifactorial, involving a multitude of triggers and substrates (Figure 1). Ectopic/triggered activity can be attributed to early afterdepolarisation (EAD), delayed afterdepolarisation (DAD) or abnormal automaticity. EAD often happens at the action potential (AP) phase 2 or 3, which is associated with AP prolongation that allows L-type Ca2+ channels to recover and activate again. In contrast, DAD is often associated with intracellular Ca2+ overload.32
There is a lack of evidence about the role of EAD in AF patients without delayed repolarisation; therefore, DAD is regarded as the major type of ectopic/triggered activity in AF arrhythmogenesis.33 Reentry refers to a phenomenon whereby the impulse travels around a specific obstacle (either anatomic or functional) and excites tissue that regains activation ability from previous excitation without extinguishing.34 Short effective repolarisation duration (ERP), slow conduction velocities as well as heterogenous electrophysiological properties are prominent factors in the formation of reentry.35,36 Often, these two factors interact and enhance each other in the development of AF. The mechanisms underlying DIAF are also diverse and may involve direct or indirect effects on atrial electrophysiology, ion channels and signalling pathways, leading to the development of initiators and/or substrates for AF. Identification of the substrates and triggers of DIAF is the first step in its management.
Drugs Affecting the Autonomic Nervous System
The pathophysiology of AF involves the critical contribution of both the vagal and sympathetic nervous systems, each with unique mechanisms.37 However, in most cases, disharmony of autonomic nervous system activation, rather than increased activity of each side respectively alone, is of greater importance for AF onset.
Sympathetic Activating Agents
Upon binding to cardiac β1-adrenergic receptors, the sympathetic neurotransmitters, catecholamines, activate adenylyl cyclase and initiate cyclic adenosine monophosphate (cAMP) synthesis. This results in the elevation of cAMP levels, which in turn activates protein kinase A (PKA). PKA activation subsequently triggers the phosphorylation of several crucial Ca2+-handling proteins. As a result of sympathetic activation, intracellular Ca2+ levels increase, which may result in Ca2+ overload and DAD.38 Catecholamines and other sympathomimetic agents are widely used as inotropes or bronchodilators, many of which can trigger AF, and dobutamine-induced AF is common in dobutamine stress echocardiography testing.39,40 Given the crucial role of the sympathetic nervous system in AF and the increased prevalence of catecholamine-induced AF, β-blockers have been widely used in the management of AF, particularly in individuals with heightened sympathetic tone.
Cholinergic Activators
ACh is the neurotransmitter released by parasympathetic efferent fibres. Vagal stimulation induces the release of ACh, which primarily binds to cardiac M2 receptors. This activation, in turn, stimulates the inward rectifier potassium current channel, IKAch, causing an outward potassium current and shortening of atrial ERP, which facilitates the formation of reentry. In addition, the non-uniform distribution of vagal nerve endings can lead to increased atrial heterogeneity, further contributing to the formation of reentry.37 Furthermore, ACh can also cause vasoconstriction in the intracoronary ACh provocation test used in the diagnosis of vasospastic angina, triggering paroxysmal AF in patients with ischaemia.20
Cardiovascular Drugs
Diuretics
In addition to their intended effects, diuretics, such as loop diuretics and thiazides, also inhibit the reabsorption of potassium ions, leading to hypokalaemia. The clinical consequences of hypokalaemia can vary, ranging from asymptomatic to potentially fatal arrhythmias.41 Current estimates suggest that up to 80% of patients receiving diuretics are at risk of developing hypokalaemia, which may lead to AF.42,43
A study by Lin et al. found that the loop diuretic furosemide was associated with an increased risk of developing AF after pacemaker implantation in elderly patients, while the thiazide diuretic hydrochlorothiazide was protective against AF in these patients.44 The researchers attributed the proarrhythmic effects of furosemide to the activation of the renin–angiotensin–aldosterone system and sympathetic nervous system, as well as hypomagnesaemia.44 Anti-aldosterone diuretics do not exhibit a significant impact on potassium levels and certain studies suggest that they may reduce the risk of AF.45
Cardiac Calcitropes
Traditional inotropes, also known as cardiac calcitropes, augment myocardial contractility by elevating Ca2+ and are commonly administered to preserve cardiac output in patients with heart failure and those undergoing post-surgical procedures. Nevertheless, heightened energy demand, as well as higher Ca2+, can lead to arrhythmias and exacerbate clinical conditions.46 Catecholamines and some sympathomimetic drugs are also important inotropes able to induce AF, as discussed earlier.
Milrinone, a phosphodiesterase 3 inhibitor, elevates cytosolic Ca2+ by preventing the hydrolysis and degradation of cyclic nucleotides cAMP/cyclic guanosine monophosphate through non-catecholamine pathways, which can promote the generation of ectopic impulses and potentially facilitate AF.11,12,47 Digitalis inhibits Na+–K+–ATPase and leads to an increase in intracellular Na+, which in turn activates the Na+/Ca2+ exchanger to promote Ca2+ influx. While atrioventricular (AV) nodal block and/or premature contractions (both atrial and ventricular) are the most frequently observed types of digitalis-induced arrhythmias, it is theoretically possible for digitalis to result in any form of arrhythmia, including AF.48,49
Vasodilators
Nicorandil, an ATP-sensitive potassium channel activator, exhibits pronounced vasodilatory effects on both coronary and peripheral vessels, rendering it a frequently used therapeutic agent in individuals with coronary artery disease. However, the activation of potassium channels also shortens atrial ERP, facilitating the formation of reentry, which plays a pivotal role in the pathophysiology of AF. Lee et al. revealed that the new application of nicorandil is linked to an augmented risk of AF/atrial flutter (OR 2.34; 95% CI [1.07–5.13]) compared with nitrate use.50 However, because of a lack of associated research, the exact incidence of nicorandil-induced AF remains unknown.
Antiarrhythmic Agents
Proarrhythmic effects remain an important issue for antiarrhythmic agents; for example, Vaughan Williams class III drugs prolong QT interval and can even cause fatal torsades de pointes.51 However, with much attention paid to ventricular arrhythmia, many antiarrhythmic drugs, such as adenosine, amiodarone and ivabradine, may also lead to or exaggerate AF.
Adenosine
Adenosine activates A1 receptors, the predominant cardiac subtype receptor, and stimulates IKAdo, an inward potassium current also activated by M2 ACh receptors.52 Consequently, adenosine-induced IKAdo current shortens AP duration (APD) and abbreviates atrial ERP in a heterogeneous manner, similar to ACh-induced atrial electrophysiological alterations that promote AF.53,54 Notably, adenosine is frequently used for the diagnosis or termination of paroxysmal supraventricular tachycardia involving the AV node, primarily because of its ability to impede AV node conduction. Nonetheless, a well-known adverse effect of adenosine is the induction of AF.17,18 While its impact is brief because of its short half-life (lasting only 20–30 seconds), this poses a significant risk, especially in patients with Wolff-Parkinson-White (WPW) syndrome, as it can potentially progress to VF and result in unstable haemodynamics.55 In such cases, the emergency department should be prepared for continuous monitoring and the availability of resuscitation equipment for the rare cases of adenosine-induced fast ventricular preexcitation in patients with WPW syndrome.55
Amiodarone
Amiodarone – used for both rate control and pharmacological cardioversion for AF patients – may also induce AF.56,57 Amiodarone, with a chemical structure resembling that of thyroid hormones, contains approximately 37% iodine. Because of its lipophilicity, slow absorption rate, moderate to low bioavailability (approximately 40–50%) and long half-life, amiodarone can accumulate in various organs and tissues, particularly with prolonged use.58 Accumulated amiodarone results in elevated iodine content, excessive hormone synthesis and release and release of stored hormone because of autoimmune destructive thyroiditis.58 As a result, chronic amiodarone administration may increase thyroid function, leading to AF. Dronedarone, developed as a non-iodinated analogue of amiodarone with similar antiarrhythmic properties, is associated with a higher rate of AF recurrence compared with amiodarone, but with fewer extracardiac adverse effects, such as thyroid dysfunction.59,60 Therefore, in selected patients (such as those with paroxysmal AF without structural heart diseases), dronedarone, but not amiodarone, may be considered the preferred choice.
Ivabradine
Ivabradine selectively inhibits the sinoatrial node (SAN) If current to decrease resting heart rate; primary clinical applications include the treatment of stable angina pectoris, left ventricular systolic dysfunction and chronic heart failure.61 However, debate remains about the relationship between ivabradine and AF.62 Ivabradine might be used to control ventricular rate in AF patients and to treat atrial tachycardia.63,64 Additionally, it shows benefits in the prevention of postoperative AF.65 There have also been various preclinical experiments supporting the anti-AF properties of ivabradine.66–69 On the contrary, other studies demonstrate evidence of elevated risks of AF by unselective ivabradine treatment in patients with chronic heart failure and coronary heart diseases, although ivabradine-induced AF did not appear to affect the incidence of non-fatal stroke compared with placebo.65,70 Laboratory studies on the underlying mechanisms are insufficient to accurately capture this complexity. It was supposed that AF might be secondary to hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 downregulation and SAN dysfunction.62 Ivabradine also leads to bradycardia, hypotension and resultant sympathetic nervous system excitation. Simultaneous vagal (i.e., heart rate-lowering) and sympathetic activation may lead to Ca2+ instability and ectopic firing.62 In a canine model, AF inducibility remained unchanged despite obvious ivabradine-induced bradycardia and vagal activity alterations.71 Overall, the inconsistent findings between studies call for further research to explore how the application of ivabradine influences AF in patients.
Anti-tumour Drugs
The use of anti-cancer drugs has been demonstrated to be an independent risk factor for AF in a dose-dependent manner.31 The underlying mechanisms of this association are still being investigated and include – but are not limited to – direct myocardial damage, modification of molecules associated with electrophysiological alterations and immune cell infiltration.
Traditional Chemotherapy
Traditional chemotherapies are typical cytotoxic drugs that target tumour cells by disrupting their metabolism or interfering with DNA replication. However, these drugs also exhibit significant cardiotoxicity and are closely associated with the development of AF.
Anthracyclines, a class of traditional antineoplastic agents, exert their anti-tumour effects by damaging DNA, generating reactive oxidative species (ROS) and inhibiting molecular synthesis. Their cardiotoxicity is cumulative, dose-dependent and augmented by risk factors such as age, pre-existing cardiovascular disease and concomitant administration of drugs with potential cardiovascular effects. While much attention is given to anthracycline-related left ventricular dysfunction, it is important to note that anthracyclines can also lead to atrial abnormalities and AF.13,14,72
In a study by Tan et al., post-surgery breast cancer patients receiving anthracycline-containing regimens showed unchanged ventricular parameters and functions before and after chemotherapy, but left atrial reservoir and conduit function were decreased post-chemotherapy.72 The authors also showed that mice treated with doxorubicin showed significant atrial remodelling and increased susceptibility to AF, which could be alleviated by dexrazoxane (a Food and Drug Administration-approved drug for management of doxorubicin cardiotoxicity) or antioxidants.72
Although the mechanisms underlying anthracycline-induced cardiotoxicity have not been fully elucidated, several pathways have been implicated, including generation of ROS, lipid peroxidation, iron-mediated damage and disruption of calcium homeostasis.73 The main mechanism of anthracycline cardiotoxicity is now thought to be through the inhibition of topoisomerase 2β, leading to the activation of the cell death pathway and concurrent inhibition of mitochondrial biogenesis.73
Novel Molecular Anti-tumour Therapies
Traditional chemotherapies – while used for many years – lack specificity toward cancer cells, causing unexpected adverse effects in normal tissues. To address this, targeted therapies have been developed to focus on molecules specific to malignant cells, aiming for higher efficacy and fewer adverse effects. However, it is important to note that even targeted therapies can lead to cardiotoxicity and heart dysfunction.
Erb-b2 Receptor Tyrosine Kinase 2 Inhibitors
Trastuzumab is a humanised monoclonal antibody that specifically targets and blocks the erb-b2 receptor tyrosine kinase 2 (ErbB2) pathway; it has become an integral part of the standard treatment of ErbB2-positive breast cancer. However, it also causes cardiac dysfunction, such as heart failure and AF, especially when used in combination with other cytotoxic drugs.19
The cardiac adverse effects of trastuzumab are attributed to blockade of the neuregulin 1 (NRG-1)/ErbB2 axis. NRG-1, released from cardiac endothelial cells in a paracrine manner, is critical in foetal cardiac tissue development and protective for adult hearts under various stimuli, e.g. stress. In cardiac tissues, there are four types of ErbB receptor, with ErbB2 being of particular importance.74 The combination of NRG-1 with ErbB receptors activates downstream effectors, such as ERK1/2 and phosphoinositide 3-kinase (PI3K)–Akt.75 ErbB signals protect the heart from various stimuli, like anthracycline treatment, possibly via anti-oxidative action.76 It has been observed that trastuzumab induced blockade of NRG-1/ErbB pathway, increased ROS production and removed its cardio-protective effect.77 ErbB2 inhibition also caused cardiac mitochondrial-associated cell apoptosis.78 All these changes eventually led to cardiac injury and predispose the heart to arrhythmias.
Ibrutinib
Ibrutinib, a first-generation Bruton’s tyrosine kinase (BTK) inhibitor, has demonstrated remarkable clinical efficacy in treating various B-cell malignancies.79 While generally well-tolerated, ibrutinib is associated with several adverse effects, with AF being one of the most common.80 Notably, among all anti-tumour agents, ibrutinib stands out as the most prominent inducer of AF.
Although the exact mechanisms underlying ibrutinib-induced AF remain unclear, it is generally considered that the blockade of cardiac off-targets, such as PI3K–Akt signalling, ErbB2 signalling and C-terminal Src kinase-Src family tyrosine kinase signalling, is responsible for its proarrhythmic effects.81–84 BTK–PI3K–Akt signalling is cardiac-protective under stress conditions and plays an important role in preventing stress-induced cardiomyopathy. Experimental evidence shows that genetically modified mice with reduced cardiac PI3K–Akt activity are more susceptible to AF, indicating a potential link between reduced PI3K–Akt activity and AF susceptibility.85 Furthermore, clinical studies have reported that patients with AF tend to exhibit lower cardiac PI3K–Akt activity levels, supporting the involvement of the pathway in the development and progression of AF.86 The proarrhythmic effect of ibrutinib was also considered to be associated with augmented late sodium current, although APD prolongation was not identified in atrial cardiomyocytes isolated from ibrutinib-treated mice.84,87 In comparison, second-generation BTK inhibitors with better selectivity for BTK have been found not to increase the risk of AF and bleeding compared with ibrutinib.88
Immunotherapies
Immune escape is an important mechanism by which cancer cells avoid being detected and eliminated; immunotherapy, which modulates the immune system to regain the ability to kill cancer cells, has been developed recently. Cancer immunotherapies – predominantly CAR-T and ICIs – elevate the host immune response targeted to neoplastic cells, which has been effectively applied clinically.89
Immune Checkpoint Inhibitors
ICIs are monoclonal antibodies designed for immune checkpoint blockade that can upregulate T-cell-related immune responses to kill cancer cells. However, systematic intensification of T-cell immune response also leads to autoimmune reaction and damage. Although those adverse effects are mild and often reversible, cardiac adverse effects can be fatal.90 The incidence of ICI-induced AF varies vastly among different studies, from 1.1% to as high as 30%.21–25 The high diversity of incidence might be because of heterogeneity among patients enrolled in studies, and because some studies were not intended to investigate the incidence of cardiovascular complications such as AF.
ICI-related cardiotoxicity and myocarditis might predispose the heart to AF.91 Expression of immune checkpoints in the heart, while low under physiological conditions, is upregulated under stimuli such as inflammation, which can protect the heart from T-cell damage.92 The use of ICIs cancels this protective effect, therefore aggravating the impact of stimuli on the heart. It has also been suggested that neoplastic cells and the myocardium might share the same antigen as the target of T-cells. ICIs cause non-specific activation of the immune system and inflammatory cell infiltration in both neoplastic and cardiac tissue, which predisposes to AF.93
Chimeric Antigen Receptor T-cell Therapy
CAR-T therapy is a novel antineoplastic treatment in which patient’s own T cells are extracted, artificially engineered in vitro to enable them to recognise tumours specifically, multiplied and finally infused back into the patient to target cancer cells.94 Clinical research has demonstrated that CAR-T therapy can induce AF.95–97 CAR-T-induced cardiotoxicity is attributed to cytokine release syndrome, i.e., general activation of the immune system and resultant hypercytokinaemia.98 Marked supraphysiological circulating inflammatory molecules such as interleukin (IL)-6 and interferon-γ can lead to fever, vasodilation, hypotension, tachycardia and multiorgan dysfunction.99 It is postulated that IL-6 is critical in CAR-T-induced cardiotoxicity.100 Because IL-6 acts as a key biomarker of AF and elevated IL-6 contributes to generation and persistence of AF, it may link systematic inflammation with AF onset, accounting for CAR-T-induced AF.101
Drugs of Abuse
Alcohol
Alcohol abuse is a global health burden associated with many diseases.102 Among these, arrhythmias – particularly AF – have a strong correlation with alcohol consumption. The term ‘holiday heart syndrome’ was first introduced in the 1970s to characterise the emergence of dysrhythmias, particularly AF, in otherwise healthy individuals following acute alcohol consumption.103 Nevertheless, the incidence of AF subsequent to binge drinking in individuals without pre-existing AF is relatively low at approximately 0.8%.27 Persistent and heavy alcohol consumption is associated with higher AF burden; Han et al. reported a 25% elevated risk of incident AF associated with a higher cumulative alcohol burden over a 4-year period and up to 47% with sustained heavy drinking during the same timeframe.28 However, there is ongoing debate regarding the association between low to moderate alcohol consumption and AF.104–106
Mechanisms underlying AF – whether induced acutely or through chronic consumption – share certain common features but also exhibit distinctions. Acute alcohol ingestion precipitates reversible and self-terminating arrhythmias, induces autonomic imbalance and diminishes atrial ERP, particularly in the pulmonary veins.107 In contrast, chronic alcohol intake frequently results in enduring atrial remodelling, structural alterations, such as left-atrial enlargement, increased left-atrial volume and reduced left-atrial mechanical performance.108–110
Tobacco and Illicit Substances
Tobacco is one of the most detrimental drug substances abused globally. A large meta-analysis reported that tobacco smoking increased the risk of developing AF by 33% compared with never smokers.111 In addition, exposure to environmental tobacco smoke has also been linked to an increased prevalence of AF, even among non-smokers.112,113 Nicotine, the primary active ingredient in tobacco, has both addictive and sympathomimetic properties, which can trigger AF by stimulating sympathetic neurotransmission and increasing sympathetic activity.114 Nicotine is also profibrotic and the fibrosis provides the arrhythmogenic substrate.115
It has been suggested that marijuana use may also contribute to the development of AF, particularly in young individuals without significant cardiac comorbidities.116,117 The exact incidence of illicit substance-induced AF is difficult to determine for several reasons. First, it can be challenging to accurately determine the consumption of illicit substances in the real world, as there may be under-reporting or non-disclosure of drug use by individuals. Second, individuals with substance use disorders may be reluctant to seek medical attention or disclose their drug use to medical providers, further complicating the identification and diagnosis of illicit substance-induced AF. Finally, individuals who use illicit substances may often use multiple drugs of abuse simultaneously, making it difficult to attribute the onset of AF to a specific drug. Marijuana smoking resulted in immediate heart rate elevation and serum norepinephrine level increase, indicating sympathetic overactivity.118 The principal active metabolite of marijuana, δ-9-tetrahydrocannabinol, inhibits cAMP formation and decreases Ca2+ influx that, in turn, results in negative cardiac inotropy, vasodilation and hypotension by binding to the cannabinoid 1 receptor.119,120 Collectively, elevated sympathetic tone by marijuana smoking may be attributed to indirect action via cannabinoid 1 receptors.
Treatment of Drug-induced AF
It is essential to conduct more extensive studies to obtain a more precise estimate of the prevalence of DIAF and to identify the underlying mechanisms and risk factors associated with this condition. Such investigations are crucial for improving our understanding of DIAF and developing effective prevention and management strategies. There is no specific expert consensus for the treatment of DIAF, and its management is primary according to the current AF guideline.6 Because DIAF often occurs during acute hospitalisation, management strategies for DIAF can also refer to the recent scientific statement put forward by the American Heart Association.121 The principal goals of managing DIAF are to optimise haemodynamics, alleviate patient symptoms and reduce the short- and long-term risks of thromboembolism. Achieving these goals requires a comprehensive approach that includes identifying and discontinuing the causative drug, assessing and managing underlying risk factors and implementing appropriate pharmacological and non-pharmacological interventions. Individualised management strategies that consider comorbidities and drug therapy are recommended for optimal management of DIAF.
The first step is to find patients with DIAF and to establish a causative relationship between the suspected drug and AF, which can be challenging. Distinguishing between DIAF and other forms of AF can be difficult, especially in patients with multiple risk factors and comorbidities. Accurately ascribing the causality of DIAF requires a thorough evaluation of the patient’s medical history, medication usage and potential underlying mechanisms of the AF. Therefore, healthcare providers need to maintain a high level of awareness of DIAF and be knowledgeable about the various drugs that can cause or exacerbate AF.
Treatment strategies may include drug dose modification or withdrawal with adequate ECG monitoring for AF recurrence. In some cases, DIAF is transient, haemodynamically stable and spontaneously reverts to sinus rhythm (e.g. adenosine), which can be considered a benign condition with a favourable prognosis. For patients who fail to spontaneously convert to sinus rhythm after drug withdrawal, additional clinical interventions to control rate and rhythm are recommended according to current guidelines for haemodynamic optimisation as well as symptom relief. Patients can switch to other drugs with similar therapeutic effects but lower AF inducibility. However, drug discontinuation must be weighed against the risk of disease progression, particularly in cancer patients in whom use of the drug may be essential because of the seriousness of the disease being treated.31 For instance, despite a relatively high incidence of AF in patients treated with ibrutinib, patients tend to be generally manageable without discontinuation of ibrutinib and attempting to mitigate the risk of AF by reducing the dose of ibrutinib may have limited effectiveness.122,123
While there are currently no reports of DIAF leading to thrombogenesis and stroke, it is theoretically possible – particularly in cases of long-lasting DIAF. As a result, it may be appropriate to use the CHA2DS2-VASc scoring system to determine if anticoagulation therapy is necessary. However, determining the need for anticoagulation in cancer patients with DIAF can be challenging, as they are at a higher risk of both thrombosis and bleeding.31 While the CHA2DS2-VASc scoring system has not been validated in patients with malignancies, given their high risk of thrombosis, anticoagulation therapy may still be recommended. Additionally, a study of ivabradine-induced AF found that it did not affect the incidence of non-fatal stroke compared with placebo, suggesting that the CHA2DS2-VASc scoring system may not be sufficient in all situations to guide clinical practice.70 Therefore, a comprehensive assessment of stroke risk in patients with DIAF should incorporate multiple factors and be tailored to individual patient characteristics.
Close monitoring and follow-up are crucial in patients with DIAF in light of the concern of AF recurrence. This involves regular clinical assessments, ECG monitoring and medication adjustments as needed. Additionally, patients with DIAF should receive education on lifestyle modifications and avoidance of drugs that may trigger AF. As the underlying mechanisms of DIAF are not fully understood, ongoing research is necessary to improve our understanding of this condition and to develop more effective prevention and management strategies. By implementing close monitoring and follow-up, healthcare providers can optimise the management of DIAF and improve patient outcomes.
Conclusion
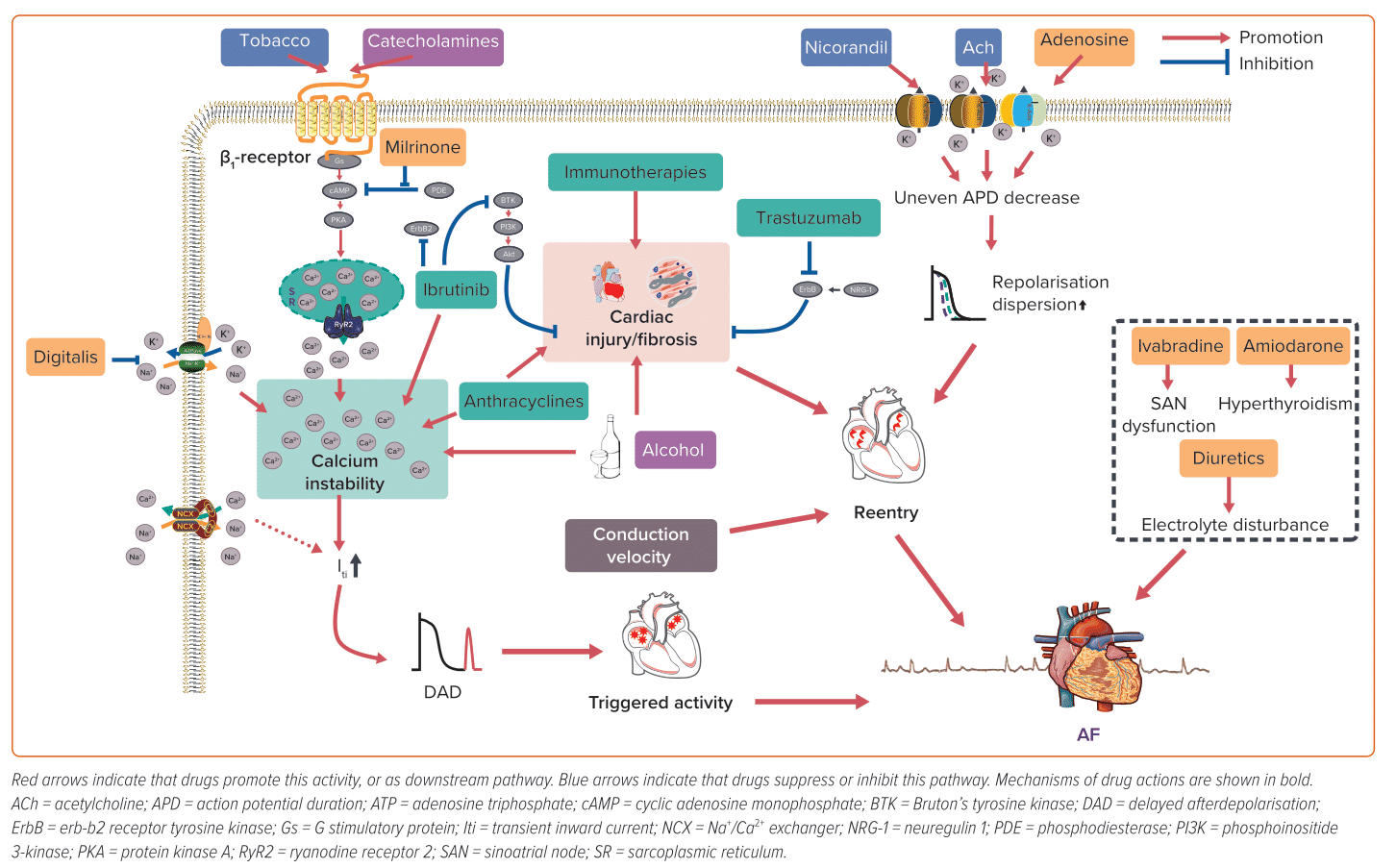
DIAF, a condition that can stem from a wide range of both cardiac and non-cardiac medications, is a prevailing clinical issue. The intricate mechanisms of DIAF include effects on ion channels, Ca2+ handling, ROS generation, inflammation and autonomic regulation of the heart (Table 2 and Figure 2). Unfortunately, DIAF is frequently overlooked and under-treated. To address this, a comprehensive understanding of the specific drug mechanisms and the individual patient’s needs and conditions are crucial for optimal management. As such, individualised approaches are recommended, with a focus on developing effective prevention and management measures. It is imperative to conduct future research investigating the underlying mechanisms of DIAF. 
Clinical Perspective
- Drug-induced AF (DIAF) is a prevalent condition that is often underestimated and overlooked and that can cause significant symptoms and outcomes.
- The management of DIAF aims to optimise haemodynamics, alleviate patient symptoms and reduce the risks of thromboembolism. Close monitoring and follow-up are crucial because of the high risks of AF recurrence. Additionally, the CHA2DS2-VASc scoring system may not be sufficient in all situations to guide clinical practice.
- Further research is needed to highlight the clinical significance of DIAF, clarify the underlying mechanisms and improve treatment strategies.