







HCN4 (hyperpolarisation-activated, cyclic nucleotide gated 4) channels, the pore-forming α–subunits of ‘funny’ channels originally described in pacemaker cells of the sinoatrial node (SAN),1 are responsible for the early phase of diastolic depolarisation in these cells and are key determinants of pacemaker generation and control of heart rate.2–5 HCN4 channels are selectively expressed in the SAN and in the conduction system, and their expression correlates tightly with the presence of spontaneous activity in adult tissue and during development; the degree of correlation is such that the HCN4 channel gene is commonly considered as a genetic marker of pacemaker tissue.6–9
The function of funny channels, and more recently of HCN4 channels, in pacemaker activity has been amply discussed in a variety of conditions.5,10 Originally described and regarded as a conceptual achievement in the understanding of the physiological basis of spontaneous activity, funny channel-based pacemaking has more recently developed into a practical notion useful in clinically relevant applications.3,11 For example, an important practical application has been the development of a family of drugs that act by specifically blocking HCN4 channels.12–14
Given the role of the funny (If) current in generation and control of rate, it is not surprising that the specific effect of these drugs is to slow the diastolic depolarisation of pacemaker cells, hence cardiac rate, with limited adverse cardiovascular side effects. Selective and quantitatively controlled slowing of heart rate provides an important therapeutic advantage in a variety of cardiac conditions. The only ‘pure’ heart rate slowing agent presently approved, ivabradine, is used as a therapeutic tool against chronic stable angina and heart failure,15,16 and off-label use in tachycardia syndromes is proving efficient.17–21
The functional properties of HCN4 channels are also the basis for the development of ‘biological’ pacemakers; the basic idea here is to induce silent or defective cardiac muscle to pace by means of gene- or cell-based in situ delivery of HCN channels. While the technology developed so far is insufficient to allow safe clinical application and replacement of electronic devices, several studies have shown that the proof-of-principle approach is feasible.22,23
A further important clinically relevant application of the concept of funny channel-based pacemaking relates to the genetic basis of arrhythmias. It is to be expected that functional defects of funny channels caused by mutations of α-subunits (HCN4) channels and/ or ancillary proteins are involved in inheritable forms of cardiac arrhythmias. Genetic screening has shown that several mutations of HCN4, and more recently also of subsidiary proteins, are associated with sinus bradycardia and/or more complex rhythm disturbances.
This short review partly extends and updates material covered by previous review publications.24-26
HCN4 Channel Gene Mutations Associated with Sinus Arrhythmias and More Complex Rhythm Disorders
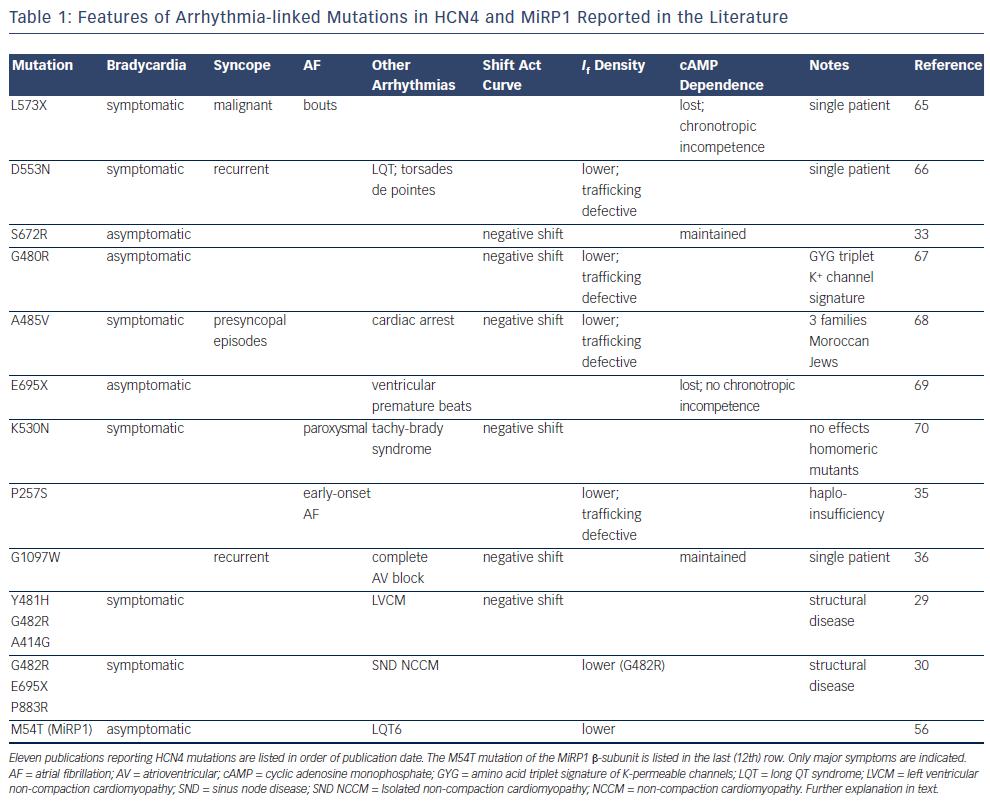
Data from genetic screening gathered in the last decade have provided substantial evidence that mutations in HCN4 are associated with rhythm disorders. A list of 11 reports, ordered according to publication date and describing HCN4 mutations in patients with sinus arrhythmias or more complex disorders is shown in Table 1. The last (12th) row of Table 1 refers to a mutation in the minK-related peptide 1 (MiRP1) protein. Other potentially harmful mutations reported in the literature for individual patients without specific investigation of genotype-phenotype association are not listed here. Some general observations highlight a few noteworthy aspects:
- All mutations are heterozygous.
- All mutations are dominant-negative, with various degrees of penetrance, except one (P257S), proposed to be dysfunctional because of haploinsufficiency.
- All are loss-of-function mutations – functional loss is caused either by a negative shift of the activation curve or by lower density of membrane expression of channels and consequent reduction of current density, typically attributable to trafficking defects or both.
- Several mutations, involving either single amino acid substitutions or truncations, are localised in the C-terminus, and three of them are in the C-linker, the region joining the sixth transmembrane domain to the cyclic nucleotide binding domain (CNBD), known to be involved in linking structural rearrangements of the CNBD to gating.27,28
- Recently reported loss-of-function mutations29,30 have been proposed to cause a symptom complex comprising bradycardia and ventricular structural abnormalities (non-compaction cardiomyopathy).
In all but two of the reported mutations, patients manifested various degrees of symptomatic or asymptomatic bradycardia. This is in accordance with the fact that mutations are loss-of-function, given the known function of the funny current in driving pacemaker activity and controlling cardiac rate. According to this concept, the size of the If current flowing during diastolic depolarisation directly determines the steepness of diastolic depolarisation itself, hence cardiac rate.31,32 For example, in the largest single family investigated so far (27 members33), the single-point mutation S672R caused, relative to wild-type channels, a negative shift of the activation curve of the If current of about 5 mV in heterozygous wild-type/mutant channels. It is interesting to note that a negative shift of the activation curve is a cholinergic-type of effect, since it mimics the inhibitory action of parasympathetic stimulation on the funny current, known to mediate vagal-induced slowing of the heart rate.2,34 Thus, the pacemaker current in patients carrying the heterozygous S672R mutation in HCN4 behaves as if in a permanent state of higher than normal vagal tone. The mutation-induced shift of the activation curve can be shown to be quantitatively adequate to slow heart rate by the amount observed within the family investigated (about 29 %).5
As well as bradycardia, other more complex arrhythmic disorders have been linked to HCN4 mutations, including ventricular premature beats, tachycardia–bradycardia syndrome and atrial fibrillation (AF), complete atrioventricular (AV) block, long QT syndrome (LQTS) and torsades de pointes. In most cases these disorders appear in addition to bradycardia, except with P257S, associated with early onset AF,35 and G1097W, associated with complete AV block.36
It is interesting to note that two recent reports29,30 provide evidence that dysfunctional HCN4 channel mutations can also be linked to cardiac structural abnormalities and specifically to non-compaction cardiomyopathy (NCCM), a disease characterised by non-compacted ventricular myocardial layer with excessive trabeculations, often associated with heart failure, arrhythmias and systemic embolic events.
These studies identify two novel loss-of-function mutations in the pore loop of the channel (Y481H and G482R) and a loss-of-function mutation in the S4–S5 linker (A414G) in patients presenting a symptom complex comprising sinus node disease and NCCM. The same combined phenotype is also reported in a family, previously investigated by the same group, carrying the HCN4-695X mutation, and in a single patient with a mutation in the terminal part of the C-terminus (P883R), though in this case no functional study is provided.30
These findings confirm the notion that, as well as controlling pacemaker activity, HCN4 channels contribute to normal cardiac development.37 Since HCN4 is expressed in cardiac progenitor cells, as a potential underlying mechanism, the authors suggest the possibility that dysfunctional HCN4 mutations directly disrupt the normal ventricular compaction process during development.29,30
The G482R mutation was found in combination with a common variant (CSRP3-W4R) in one study30 but not in another,29 suggesting that the variant is not essential, though it can act as a predisposing condition. Expression of heterozygous wild-type/G482R mutated channels generated apparently contrasting results in the two studies: a strong negative shift of the activation curve in one study29 and a reduced membrane expression with no shift of the activation curve in the other.30 In both cases, however, the changes lead to loss of function and are compatible with the bradycardic phenotype.
While, as discussed above, it is expected that a loss-of-function modification of HCN4 causes sinus bradycardia, it is less immediately obvious how an HCN4-reduced contribution to activity can correlate with AV block or AF. Interestingly, however, as discussed below, several data indicate the potential involvement of a dysfunctional funny current in AV block and AF.
It is known from early studies that funny channels are expressed in the AV node, where they contribute to spontaneous activity.38,39 Other more recent studies have identified pacing cells in the sleeves of pulmonary veins (PV), which express funny channels.40,41 Such PV cells can contribute to ectopic beat generation and thus represent important focal sources potentially able to initiate AF.42,43 A correlation between AF and expression of funny channels in the SAN has already been proposed in a study that investigated the SAN dysfunction associated with AF, normally apparent after AF termination (tachycardia–bradycardia syndrome).44 This study showed that the SAN dysfunction has a reversible component, related to the SAN remodelling caused by rapid atrial tachyarrhythmias, which involves If downregulation. Furthermore, HCN4 was identified more recently as a candidate AF-linked gene in a large-scale meta-analysis of genome-wide association studies (GWAS) conducted to detect new AF susceptibility loci.45
In the study of Macri et al.35 the authors did not investigate specific families but rather collected early onset AF patients from the Massachusetts General Hospital (MGH) AF study and compared them with controls from the Framingham Heart Study (FHS). Sequencing for HCN4 variants led to the identification of several single nucleotide polymorphisms (SNPs) with a higher rate of expression in AF patients. Only one of these variants, P257S, was functionally different from wild-type, and expression in Chinese hamster ovary (CHO) cells of mutated HCN4 channels resulted in no current, indicating that P257S channels did not traffic to the cell membrane. This is consistent with the idea that the N-terminal region of HCN4 channels is involved in membrane insertion of the protein.46 Curiously, however, the loss-offunction property was lost when the mutated protein was expressed in heterozygous conditions, excluding a dominant-negative effect of the mutation. The authors interpreted these data to indicate that early-onset AF in P257S carriers is due to haploinsufficiency, i.e. only wild-type proteins contributing to functional channels. It should be noted in the context that the P257 residue is part of the caveolinbinding sequence present in the N-terminus of HCN4 channels47 and that a mutation-induced disruption of the channel binding to caveolin is expected to cause reduced membrane expression.
In relation to AV block, indication of a potential contribution of defective HCN4 is apparent, for example from HCN4 knockout studies. In a study investigating inducible, cardiac specific knockout of HCN4 channels, Baruscotti et al.48 found that HCN4 knockout causes, as expected, progressive slowing of sinus rate. This process was, however, accompanied by a progressive prolongation of the PQ interval that evolved into second-degree block and eventually complete AV block and heart arrest in knockout animals, suggesting a role of HCN4 channels in AV node (AVN) conduction. Interestingly, a similar conclusion was suggested by experiments showing that block of the funny current generates a larger response in AVN than in SAN myocytes.39
In the study of Zhou et al.,36 a single patient with complete AV block, but no sinus node dysfunction, who had undergone pacemaker implantation, was found to carry the HCN4 mutation G1097W. This was shown to be a loss-of-function mutation associated with a negative shift of the activation curve and a lower membrane expression level. It is worth noting that before pacemaker implantation the patient had a 4:1 conduction ratio, with a sinus rate of 132 and a ventricular rate of 33 bpm, suggesting the presence of a reflex sinus tachycardia compensating for the ventricular slow rate. This is interesting since it suggests that a basal sinus bradycardia, hidden by a reflex sinus tachycardia, cannot be excluded.
Screening of Defective HCN4 Auxiliary Subunits – MiRP1
The results discussed above indicate that several types of arrhythmias, some of which complex, are found in patients with HCN4 mutations, yet all mutations are loss-of-function and only affect either channel kinetics (by shifting the activation range of the current to more negative voltages), or membrane expression (by decreasing it), or both. Clearly, changes in the HCN4 channel only cannot explain the whole variety of rhythm disorders found, and other factors are likely to contribute.
In line with this consideration, it should be taken into account that HCN4 channels represent only one of the components of funny channels (i.e. the α-subunits), and that disturbances of the pacemaker process can also derive from alterations of auxiliary subunits or interfering proteins (such as for example MiRP149,50 or caveolin-347,51,52) or other proteins such as KCR1 and SAP97.53 Screening of these proteins in arrhythmia patients can thus provide useful insight into new potentially causative mutations.
One published example is mutation M54T of the MiRP1 β-subunit (KCNE2 gene). MiRP1 is a hERG potassium channel β-subunit,54 which also interacts with HCN channels.50 Mutations of MiRP1 that reduce delayed potassium currents have been reported in LQTS patients, and are thought to be responsible for QT prolongation and delayed repolarisation.54–56
Sinus node dysfunction and bradycardia are sometimes observed in LQTS patients, and to investigate if the bradycardia associated with LQTS involves changes in the funny current, Nawathe et al.56 analysed the effects on HCN channels of the MiRP1 M54T mutation found in a patient from an LQTS6 registry with bradycardia (see last row in Table 1). Functional studies performed by co-expressing wild-type or mutated MiRP1 subunits with HCN4 showed that the M54T mutation strongly decreases the HCN4 current contribution to activity. Numerical reconstruction obtained by combining changes induced in HCN4 and hERG potassium channels by the M54T MiRP1 mutation was able to mimic bradycardia. These data represent the first evidence that the contribution of funny channels to activity can be altered and become arrhythmogenic in the presence of dysfunctional auxiliary subunits.
Conclusions and Future Perspectives
A direct way to appreciate the relevance of funny/HCN4 channels to pacemaking is to look at the consequences of their modifications on rhythmic activity. There are several ways by which the normal contribution of the If current to activity can be altered.
Alteration of normal function is primarily caused by gene mutations, which, as discussed above, can occur in the channel α-subunits as well as in auxiliary subunits like MiRP1, caveolin-3 and other elements known to interact with funny channels.53 Clearly, in this latter case other channels may also undergo modifications since auxiliary subunits often regulate multiple targets. HCN4 mutations found so far are all loss-of-function, and it will be interesting to see if gain-of-function mutations also occur. Based on the established role of the funny current, it is to be expected that these mutations are associated with tachyarrhythmias.
Finally, another way by which the channels normal function can be altered is remodelling. In general, remodelling can be viewed as the set of processes by which channel activity is regulated, via turnover of membrane expression, by DNA transcriptional control. While this mechanism is normally physiological, it can be modified by non-physiological conditions and under specific circumstances lead to abnormal contribution of ion channels to activity.
Funny channels undergo remodelling and altered contribution in various cardiac diseases. It is known for example that funny channels are overexpressed in the ventricular muscle of human failing hearts in dilated cardiomyopathy, and that this mechanism is potentially pro-arrhythmogenic.57,58 As mentioned above, downregulation of HCN channels has been reported in tachycardia-induced SAN remodeling.44 Also, remodelling of the peripheral cardiac conduction system in response to pressure overload causes hypertrophy and a four-fold increase in the expression of HCN4.59
A more recent study has investigated the effects of training on cardiac ion channel remodelling. It is generally assumed that exercise training-induced bradycardia is caused by an increased parasympathetic drive.60,61 This view has been recently challenged by a novel, radically different interpretation. A collaborative study by D’Souza et al.62 has shown that slowing of heart rate induced by exercise training in rodents is actually caused by downregulation of HCN4, rather than to increased parasympathetic activity. HCN4 channel downregulation has a major role in determining heart rate, but it occurs in the framework of a widespread remodelling involving ion channels, transporters, transcriptional factors and other molecules. This finding may explain why endurance athletes are often subject to bradycardia associated with a higher incidence of sinus node disease and require electronic pacemaker implantation more frequently than sedentary patients.63,64
It is likely that more mechanisms involving modification of funny channel contribution to pacemaking, including gene mutations of α or ancillary subunits, changes of channel modulation processes, interacting proteins, remodelling of ion channels and related proteins, will need to be added in the near future to those listed here.