







Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a rare heritable cardiomyopathy characterised by fibro-fatty replacement of the myocardium, which predisposes patients to frequent lifethreatening ventricular arrhythmias and slowly progressive ventricular dysfunction.1,2 Structural involvement of the right ventricle (RV) generally predominates,3,4 although left dominant forms of ARVD/C are increasingly well-recognised.5 Patients typically present in their second to fifth decade with symptoms associated with ventricular arrhythmias and go on to have an arrhythmic disease course.6,7 Sudden cardiac death may be the first manifestation in up to 50 % of index cases.8
ARVD/C was first described in the modern scientific literature in 1982 in the seminal work of Frank Marcus and colleagues.9 In this initial description of 24 patients with ventricular arrhythmias and RV dysfunction from a French tertiary care centre, the authors speculated that ARVD/C resulted from a developmental abnormality of the RV musculature. Indeed, the original name – arrhythmogenic RV dysplasia – reflects this early interpretation. However, soon afterwards, threads of evidence implicating both inherited factors (nature) and exercise (nurture) in ARVD/C pathogenesis emerged. Clustering of ARVD/C within families was appreciated early.10 Investigators recognised the cardiac phenotype of a rare familial cardio-cutaneous condition, Naxos disease, overlapped with ARVD/C.11 The major discovery that homozygous mutations in junctional plakoglobin (JUP) were the genetic basis of Naxos disease in 2000,12 led to rapid identification of mutations in each of the desmosomal genes among ARVD/C patients. Simultaneously, articles began appearing calling attention to the fact that many patients with ARVD/C were elite athletes13 and that athletic ARVD/C patients appeared to have a particularly high risk of sudden cardiac death.14,15
These early observations set the stage for research exploring the genetic basis of ARVD/C (nature) and the role of endurance exercise (nurture) in ARVD/C pathogenesis and clinical course. This article will review current evidence regarding the association of genotype, endurance exercise and clinical phenotype of ARVD/C patients and at-risk family members and discuss the emerging paradigm in which genetic predisposition and environmental factors (exercise) interact around a threshold for phenotypic expression of ARVD/C. Excellent reviews have recently been published describing current understanding of the molecular mechanism through which ARVD/C-associated mutations lead to disease.16,17 Thus, this paper will focus on evidence from clinical research and address three questions: 1) To what extent does genotype predict ARVD/C phenotype? 2) How is endurance exercise associated with ARVD/C phenotype and clinical course? 3) How do exercise and genotype interact in disease pathogenesis?
Nature – To what Extent does Genotype Predict ARVD/C Phenotype?
Genetic Basis of ARVD/C
Following the landmark discovery in 2000 that mutations in JUP, which encodes plakoglobin, was the cause of Naxos disease,12 there was rapid discovery of ARVD/C-associated mutations in each of the desmosomal genes including DSP encoding desmoplakin,18 PKP2 encoding plakophilin-2,19 DSG2 encoding desmoglein-220 and DSC2 encoding desmocollin-2.21 Cardiac desmosomes are specialised adhesion junctions composed of a symmetrical group of proteins – the cadherins, the armadillo proteins and the plakins – that provide a mechanical connection between cardiac myocytes. It is now known that up to two-thirds of ARVD/C patients have mutations in genes encoding the cardiac desmosome,22 with heterozygous radical mutations in PKP2 most prevalent among the North American and most European populations.22,23 Inheritance of desmosomal mutations typically follows an autosomal dominant pattern with incomplete penetrance and variable expressivity. However, patients with multiple mutations (compound heterozygosity and digenic) are not uncommon.24 Cases with homozygous mutations and pedigrees more reminiscent of autosomal recessive disease also occur.25,26 The reported proportion of ARVD/C patients with multiple mutations ranges widely (4–21 %)23,27 and is likely related to how stringently missense variants are adjudicated.28
Although the vast majority of reported ARVD/C-associated mutations are desmosomal (95.5 % of mutations reported in the ARVD/C Genetic Variant Database),29 extradesmosomal mutations are being identified in an increasing minority of patients. The first of these was a founder mutation in transmembrane protein 43 (TMEM43) S358L discovered among families in Newfoundland, Canada.30 More recently, mutations have been reported in genes previously associated with other cardiomyopathies and arrhythmia syndromes including desmin (DES),31 titin (TTN),32 lamin A/C (LMNA),33 phospholamban (PLN)34 and Nav1.5 (SCN5A),35 the pore-forming subunit of the voltage-gated cardiac sodium channel. These findings likely reflect clinical overlap of ARVD/C with dilated cardiomyopathy at one phenotypic extreme36 and with arrhythmia syndromes associated with sodium channel dysfunction, particularly Brugada syndrome, at the other.17 Mutations in the CTNNA3 gene coding for α T-catenin protein in the ‘area composita’, (composed of both desmosomal and adherens junctional proteins), have also recently been shown to be associated with ARVD/C.37 Finally, mutations in transforming growth factor β3 (TGFβ3)38 and the cardiac ryanodine receptor-2 (RYR2)39 have been described, although the association of mutations in these genes with an ARVD/C phenotype have not been confirmed.
The increasing recognition of mutations in extradesmosomal genes among ARVD/C patients is largely the result of advances in geneticsequencing technology (next-generation sequencing). This technology allows for comprehensive sequencing at low cost with rapid turnaround. While identification of mutations in novel ARVD/C genes is useful for both scientific discovery and clinical management, it comes with challenges associated with interpretation of sequence results. As increasing numbers of genes are sequenced in ARVD/C patients, increasing numbers of genetic variants with uncertain pathogenicity are revealed. Missense variants are a significant interpretive challenge. It is important to realise that assessing pathogenicity of genetic variants should be performed with knowledge of the background ‘genetic noise’ in a healthy population. A study by Kapplinger et al.40 focusing only on the desmosomal genes highlights the complexity. In this study of 427 putatively healthy individuals, 16 % were found to carry a rare missense variant in the desmosomal genes. (In this study the authors considered each variant exclusively observed in ARVD/C patients but absent in a large, ethnically matched, control cohort a ‘mutation’.) A recently updated ARVD/C-specific genetic variant database (http://www.arvcdatabase.info) encompassing more than 1,400 variants collating published evidence is useful for interpretation.29 Recent recommendations for adjudication of genetic variants suggest increasingly stringent criteria be used.41
Finally, there remain many ARVD/C cases with no identifiable mutation. In the largest study of ARVD/C families to date, among 439 index cases ascertained through the Johns Hopkins and Dutch Interuniversity Cardiology Institute of the Netherlands (ICIN) ARVD/C Registries, 37 % had no identifiable mutation in the desmosomal genes, PLN or TMEM43.7 Of interest, among these cases without mutations, clear evidence of familial disease (meeting ARVD/C 2010 Family History Task Force Criteria)42 was present in only one-fifth. This raises the question of whether the remaining 80 % of cases had ARVD/C caused primarily by genetic factors, a question we will return to in the latter part of the paper.
Association of Genotype with Clinical Characteristics
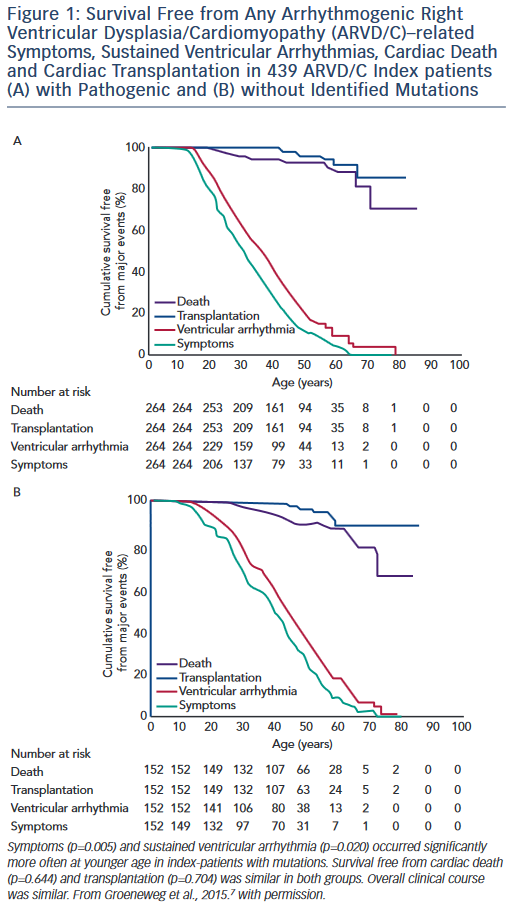
Studies of the association of genotype with clinical ARVD/C phenotype are challenging due to broad clinical variability even within families who share both genotype and environment. However, clinically useful patterns of genotype/phenotype associations have begun to emerge. Broadly, the clinical course of ARVD/C does not differ substantially between ARVD/C index patients with and without identifiable mutations.7 As shown in Figure 1, regardless of mutation status, ARVD/C is a highly arrhythmic condition. Most index patients experience symptoms and sustained ventricular arrhythmias, but only a minority heart failure or transplant. The single exception to this pattern of overall similarity was that index patients with mutations had a significantly younger age of symptom onset and first ventricular arrhythmias than patients without mutations. This pattern of earlier onset is commonly seen when comparing strongly genetic forms of diseases with those with complex multifactorial inheritance.
In a pattern also observed among many genetic diseases, several groups of investigators have found that ARVD/C patients with more than one desmosomal mutation have worse phenotypes than patients with a single or no mutation. Rigato et al. observed increased penetrance and worse arrhythmic outcomes in 21 Italian carriers of multiple mutations in comparison to 113 carriers of single-desmosomal mutations.43 They showed that being a carrier of more than one mutation was the most significant risk factor for malignant arrhythmic events and sudden death in their population. In another study, a higher incidence of cardiogenic syncopal events occurred in multiple mutations carriers among a Chinese population of ARVD/C patients.27 Finally, in an analysis of 577 desmosomal, PLN and TMEM43 mutation carriers drawn from the Johns Hopkins and Dutch ARVD/C Registries, the 4 % of patients with multiple mutations had significantly earlier occurrence of sustained ventricular tachycardia/ventricular fibrillation, a lower rate of survival free from sustained ventricular arrhythmias and more frequent left ventricular (LV) dysfunction, Class C heart failure and cardiac transplantation.23 Taken together, these data suggest an effect of gene dosage on ARVD/C phenotype. Based on these data, more aggressive management of carriers of multiple mutations is indicated.
There are also several clinically relevant associations between mutations in specific desmosomal genes and ARVD/C characteristics. A higher prevalence of LV involvement occurs among carriers of DSP mutations. This association was initially recognised by Norman et al. among a British population of ARVD/C patients in 2005.44 Defects in the C-terminal of the protein were associated with early and predominant involvement of the LV and a high occurrence of sudden death.45 In a large study by Bhonsale et al.,23 DSP carriers experienced more than fourfold more LV dysfunction (40 %) and Class C heart failure (13 %) than their large group of PKP2 carriers. LV involvement is also more likely among cases with PLN mutations. (Most PLN carriers have the Dutch founder mutation c.40_42delAGA that has been identified in 13 % of ARVD/C index patients in the Netherlands).1 The TMEM43 S358L founder mutation most prevalent in Newfoundland is associated with very high disease penetrance and arrhythmic risk among male carriers in comparison to other ARVD/C patients.46
Incomplete Penetrance and Variable Expressivity
While improved understanding of the genetic basis of ARVD/C has been helpful in diagnosing and managing patients, familial ARVD/C remains a clinically heterogeneous disorder characterised by incomplete age-related penetrance and significant variable expressivity.47 With the expansion of genetic testing for ARVD/C and integration of genetic test results into the diagnostic criteria,42 clinicians are increasingly confronted with caring for at-risk mutation carriers. While the literature reports up to 83 % penetrance of desmosmal mutations (in Spanish carriers of a DSP c1339C>T mutation),48 this likely is an overestimate, or at least a ceiling. Families initially ascertained and enrolled in genetic research will have higher than typical penetrance as this is what makes them attractive for genetic studies. In the large report by Bhonsale et al. of over 500 desmosomal mutation carriers,23 roughly one-third met ARVD/C 2010 Diagnostic Task Force Criteria (TFC). When family members are diagnosed, they often have a milder course than probands.7 While the full explanation for this phenotypic heterogenity remains unclear, there is increasing evidence that exercise plays a major role in disease penetrance and arrhythmic risk.
Nurture – How is Exercise Associated with Disease Onset and Clinical Course?
Influence of Exercise on Disease Expression
The appreciation of the association of exercise and ARVD/C began with the twin observations that ARVD/C patients were frequently athletes and that arrhythmias occurred disproportionately during athletic activity in ARVD/C patients.13,15 A review of autopsies in the Veneto region of Italy showed participation in competitive athletics resulted in a more than fivefold increase in sudden cardiac death risk among young patients with ARVD/C.14 Consistent with this observation, implementation of a pre-participation screening programme resulted in a sharp decline in such deaths.49 The first observation that athletic involvement correlated with disease characteristics among living patients was by Sen-Chowdhry and colleagues who found that among a group of 116 ARVD/C patients, the 11 patients who participated in long-term endurance training had more severe RV dysfunction.45
During the past 2 years, research has accelerated with four clinical studies focused on addressing the extent to which exercise influences ARVD/C onset and disease course. These studies, reviewed below, make a compelling case that there is a dose-dependent relationship between exercise intensity and duration and disease severity.
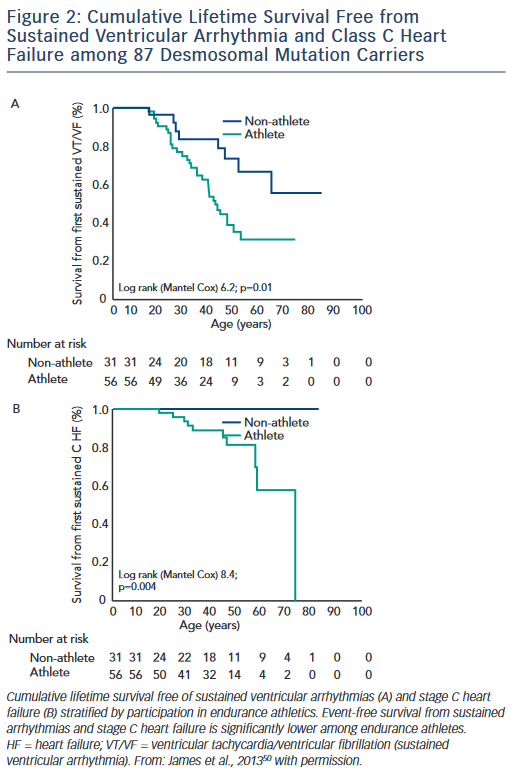
The first study, published by James et al.,50 collected exercise history by interview from 87 carriers of heterozygous desmosomal mutations ascertained from the Johns Hopkins ARVD/C Registry. Endurance athletes were defined as participants in a sport with a high dynamic demand (>70 % Max O2), based on the 36th Bethesda Conference Classification of Sports (Task Force 8),51 for at least 50 hours/year at vigorous intensity. The results of the study showed that both participation in vigorous endurance (aerobic) athletics and greater duration of annual exercise were associated with an increased likelihood of ARVD/C diagnosis in a dose-dependent fashion. Similarly, as shown in Figure 2, survival from first sustained ventricular arrhythmia and onset of Class C heart failure was worse among athletes. Furthermore, the study suggested that modifications in exercise following clinical presentation could alter clinical course. Mutation carriers who continued to participate in high duration exercise after clinical presentation had worse survival from first ventricular arrhythmia compared with individuals who reduced their exercise. Taken together these data suggested an important link between exercise and the outcomes of desmosomal mutation positive ARVD/C patients and family members.
These findings were subsequently confirmed in both European and multicentre North American populations and in ARVD/C patients without desmosomal mutations. Saberniak and colleagues investigated the impact of exercise on myocardial function among 110 Scandinavian ARVD/C patients (both with and without desmosomal mutations) and mutation carrier family members.22 Data were again collected by interview. Exercise participation was measured by metabolic equivalent (MET)-minutes per week with patients participating in vigorous (≥6 METs) physical activity for ≥4 hours/week classified as athletes. The authors showed that athletes were more likely to meet diagnostic criteria, experience ventricular arrhythmias and progress to cardiac transplant. Furthermore, they found that both RV and LV function were significantly reduced in athletes and that intensity of exercise was correlated with the extent of structural dysfunction in a dose-dependent fashion.
Next, the Johns Hopkins group confirmed the association of exercise with clinical course among 43 ARVD/C index patients without desmosomal, PLN or TMEM43 mutations. Patients were categorised as athletes using the same criteria previously described. Sawant et al.52 found that those participating in the highest intensity (MET-hours/ year) exercise prior to clinical presentation had earlier onset, worse RV structural abnormalities and poorer survival from ventricular arrhythmias in follow-up.
Finally, Ruwald et al. recently reported data from 108 probands ascertained in the North American ARVC registry.53 For this study patients self-reported whether they were inactive, recreational athletes or competitive athletes both prior to diagnosis and in follow-up. Similar to the findings of James et al., Sawant et al. and Saberniak et al., competitive athletes had a worse clinical profile with earlier symptom onset, and a worse survival from a combined ventricular tachyarrhythmia/death endpoint. They also noted that individuals who continued competitive exercise had a significantly worse arrhythmic course following diagnosis compared with those who reduced exercise, validating the initial findings of James et al.50 By contrast, the authors detected no differences in age of onset or risk of ventricular arrhythmias/death between patients who rated themselves as inactive versus recreational athletes. While these data are somewhat reassuring, some caution is warranted as close examination of the data shows recreational athletes had somewhat worse LV function than inactive patients. Additionally, age of symptom onset and survival from sustained ventricular arrhythmia among recreational athletes appears to be midway between the competitive athletes and inactive patients, albeit not significantly different.
Recent studies from model systems have also implicated exercise in ARVD/C pathogenesis. Murine ARVD/C models with abnormal expression of two different desmosomal proteins (plakoglobin and plakophilin-2)54,55 have both shown evidence of earlier development of disease and worse structural abnormalities when exposed to endurance training. The molecular mechanism of this process remains unclear. Recent evidence suggests while reduction of desmosomal protein expression causes decreased cell–cell adhesion, expression of mutant protein results in cells with typical mechanical properties but abnormal signalling responses to stress.56
Implications for Exercise Recommendations for ARVD/C Patients and Family Members
Taken together these data convincingly argue that participation in vigorous or competitive endurance exercise leads to poor outcomes in ARVD/C patients. A recent international expert consensus statement on the treatment of ARVD/C57 integrated these data and recommended that ARVD/C patients be restricted from competitive and/or endurance sports (Class I recommendation). Restriction from other athletic activities with the possible exception of recreational low-intensity sports was also recommended (Class IIa). These recommendations are generally concordant with existing professional recommendations from Europe and North America.58–60
ARVD/C Pathogenesis – How do Exercise and Genotype Interact in Disease Pathogenesis?
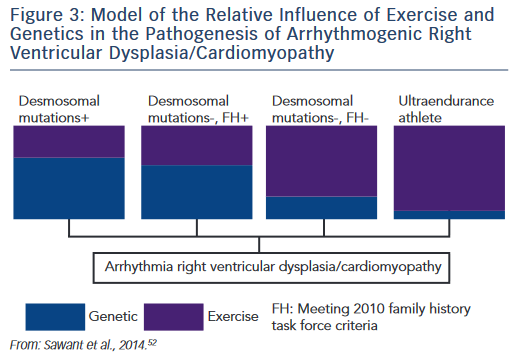
The research described establishes the role of both genetic predisposition and exercise in ARVD/C pathogenesis and course. Our understanding of how these factors interact remains incomplete. An emerging paradigm suggests there is a threshold for phenotypic expression of ARVD/C with the relative amount of exercise necessary to reach the threshold varying based on genotype.52,61 As shown in the schematic (see Figure 3), we hypothesise that individuals born with very high genetic risk, such as carriers of multiple mutations, require little (or perhaps no) exercise to promote ARVD/C onset. At the other end of the spectrum, a series of studies by Heidbüchel et al. suggests ultra-endurance athletes exposed to massive amounts of exercise may develop a predominantly exercise-induced form of ARVD/C.62 This hypothesis, first proposed in 2003,63 was developed based on a clinical pattern of a high prevalence of RV arrhythmias and predominant RV dysfunction among high-level endurance athletes referred for evaluation of palpitations and other arrhythmia-associated symptoms. Further support for this concept came from their observation that among 41 athletes with definite or probable ARVD/C, only six had a definite or possible desmosomal mutation and few had a family history of disease.64 Furthermore, their mutation carriers had done significantly less exercise than the remaining cases suggesting less exercise was required for disease onset in the setting of a genetic predisposition.
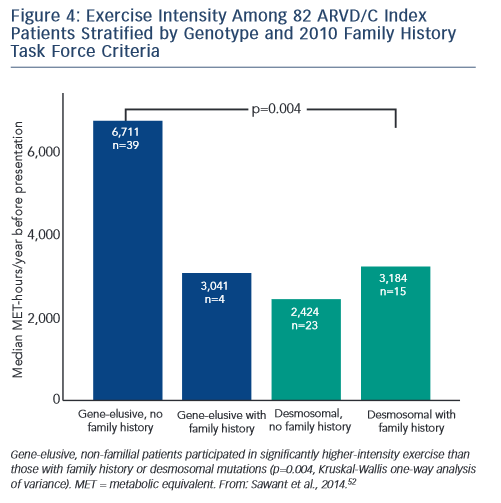
Sawant et al. recently confirmed and extended these findings via a study of 82 index cases, half of whom carried a desmosomal mutation.52 All the patients without desmosomal mutations were athletes (≥50 hours/year participation in a sport with high dynamic demand at vigorous intensity) in comparison to two-thirds of mutation carriers. Additionally, similar to the findings of Heidbüchel and colleagues, the patients without a desmosomal mutation had done considerably more intense exercise prior to clinical presentation. Sawant also found a relatively low prevalence of familial disease among cases without desmosomal mutations. Furthermore, as presented in Figure 4, gene-elusive cases with familial disease had performed exercise indistinguishable from that of desmosomal mutation carriers while the ARVD/C patients with neither a mutation nor a family history had done by far the most intense exercise.
While these studies suggest exercise plays a disproportionate role in the pathogenesis of ARVD/C cases without an apparent genetic predisposition, it is premature to conclude that this group of ARVD/C patients has an entirely acquired disease. In the study by Sawant et al., 10 % of cases with no identifiable mutation in the desmosomal genes, PLN or TMEM43 had clear evidence of familial disease by TFC. Second, ARVD/C is a rare disease (prevalence is estimated at 1/5,000)2 suggesting only a relatively small proportion of athletes are susceptible. One could speculate that this susceptibility stems from mutations in novel genes with lower penetrance or by combinations of rather low penetrant variants in both desmosomal and other genes. Other environmental factors as well as patient sex may also play a role.
Is there a Threshold for Safe Exercise for Desmosomal Mutation Carriers?
If, as the model suggests, exercise and genetic predisposition have an additive effect towards a threshold for ARVD/C pathogenesis, there may be a level of exercise at least some at-risk mutation carriers can participate in without triggering ARVD/C onset. Based on their finding of a dose–response relationship of exercise with the extent of LV and RV dysfunction, Saberniak and colleagues22 postulated that there may be no threshold value for recommendations for physical activity in at-risk mutation carriers to prevent negative effects on myocardial function. However, given the indisputable benefits of exercise for overall health, complete exercise restriction in young, otherwise healthy mutation carriers is not without risk. Unfortunately, there are few clinical recommendations for appropriate exercise for at-risk carriers. The recent ARVD/C treatment consensus statement concludes only that restriction from competitive sports may be considered in healthy gene carriers (Class IIa).57
Therefore, we recently completed a study65 assessing a threshold for disease onset based on the American Heart Association (AHA) minimum recommended exercise (450–750 MET-minutes weekly)66 for maintenance of overall health in adults. In this study we interviewed members of families segregating heterozygous radical mutations in plakophilin-2. This study design allowed us to control for genotype and other genetic and environmental variables clustering in families as the interaction of exercise and genetic predisposition likely differs based on genotype.
Unsurprisingly, probands had done more exercise than family members and athletic family members had poor outcomes. However, family members who restricted exercise at or below the upper bound of the AHA minimum were significantly less likely to be diagnosed and had no sustained ventricular arrhythmias. (These family members were not sedentary; their median exercise was 80 % of the lower AHA bound.) Furthermore, when family members were diagnosed and had a first sustained ventricular arrhythmia they had accumulated 2.8- and 3.5-fold, respectively, greater MET-hours exercise (from age 10) than the AHA-recommended minimum. By contrast, median exercise for family members who did not develop disease was close to the AHA recommended levels across the lifespan.
These results suggest that at least for many unaffected PKP2 carriers, the AHA-recommended minimum exercise level may fall below the threshold required to promote disease onset. This points to restricting these carriers from endurance and high-intensity athletics, but potentially not from AHA-minimum recommended levels of exercise for healthy adults. It is likely the threshold for healthy versus risky exercise will differ based on genotype and will almost certainly be much lower for carriers of multiple mutations. The findings also support the concept of the additive model of genetic predisposition and exercise intensity in ARVD/C pathogenesis. Future research to improve understanding of the interaction of genotype and exercise ‘dose’ as well as other environmental factors in triggering disease onset is key to improving care for families affected with ARVD/C. Additionally, improved understanding of the molecular mechanism through which exercise interacts with expression of abnormal protein or reduced protein expression to cause the pathological features of ARVD/C is critical.
Clinical Perspective
- Carriers of multiple mutations typically have a more severe ARVD/C phenotype with more frequent and earlier onset arrhythmias, structural dysfunction, heart failure and need for transplant.
- DSP and PLN carriers are at significant risk of developing LV dysfunction. Their risk is significantly higher than that of the large proportion of ARVD/C patients with PKP2 mutations for whom the risk is relatively low.
- The data support restricting ARVD/C patients from competitive, frequent, high-intensity exercise regardless of genotype.
- Unaffected PKP2 carriers of heterozygous mutations may be able to participate in exercise based on AHA-minimum guidelines for exercise in healthy adults, thus avoiding risks of a sedentary lifestyle so long as they are complying with regular ongoing clinical follow-up to detect any early signs of ARVD/C onset.