







Anticoagulation with vitamin K antagonists (VKAs) has been used for the long-term treatment and prevention of thromboembolic diseases and for stroke prevention in atrial fibrillation (AF) for the past half century. Until the last decade, VKAs were the only oral anticoagulant (OAC) agents available, and warfarin remains the most commonly prescribed OAC worldwide.1 Direct oral anticoagulants (DOACs), which selectively block key factors in the coagulation cascade, provide an effective and safe alternative to VKAs for the long-term treatment and prevention of thromboembolic diseases and for stroke prevention in AF. One of the greatest advantages of DOACs in long-term treatment is the lack of need for routine monitoring of coagulation.
However, in order to balance the imminent risk of recurrent ischaemic events against the bleeding risk, factors that impact pharmacokinetics and dynamics should be taken into account in the management of OAC therapy. Given the long-term use of DOACs, the frequent use of over-the-counter medications and the need for multiple drug treatments in patients with comorbidities, the evaluation of drug–drug interactions (DDIs) with DOACs is essential.
The aim of this article is to present information about factors that influence the activity of OACs, and interactions between OACs and genetic and other factors, such as medicines, food, diseases and pre-existing conditions. While clinical trial data for most alleged interactions are lacking, clinicians (and patients) should be aware of potential DDIs and drug–food interactions.
Vitamin K Antagonists
By targeting vitamin K epoxide reductase, the post-translational modification of the vitamin K-dependent blood-coagulation proteins is impaired, see Figure 1.2 A reduced functional level of factor IX, factor VII, factor X and prothrombin leads to delayed blood coagulation. This inhibition is monitored in the clinical laboratory with the use of prothrombin time and is corrected for variable potencies of tissue factor used in the assay by means of a calibration factor, yielding the international normalised ratio (INR).3 The goal of therapy is to keep the INR within the therapeutic range.4,5 Patients with an average individual time >70 % are within the therapeutic range and are considered to be at a low risk of a major haemorrhagic or thrombotic event.6
Although effective under optimal conditions, given the narrow therapeutic window, numerous environmental (e.g. food and drug) and genetic interactions, e.g. cytochrome P450 family 2 subfamily C member 9 (CYP2C9) or vitamin K epOxide reductase complex (VKORC)1, complicate the long-term use of these drugs and render treatment with these agents complicated.7–13
Warfarin binds to albumin, and only about 3 % is free and pharmacologically active. A number of medications (e.g. ibuprofen, losartan, valsartan, amlodipine and quinidine) can displace warfarin binding, leading to its increased activity and subsequent increased rate of degradation.14

DDIs affecting the pharmacokinetics of warfarin mainly involve inhibition of the expression and/or activity of cytochrome P450 (CYP) isoenzymes involved in warfarin metabolism (CYP3A4 for the R-enantiomer and CYP2C9 for the three- to five-times more potent S-enantiomer of warfarin).15 The concomitant use of medications that induce CYP2C9 results in increased clearance of warfarin and less anticoagulation, see Table 1. The most pertinent DDIs are with azole antifungals, macrolides, quinolones, non-steroidal anti-inflammatory drugs (including selective cyclooxygenase-2 inhibitors), selective serotonin reuptake inhibitors, omeprazole, statins, amiodarone and fluorouracil.14 In the Apixaban for Reduction In Stroke and Other Thromboembolic Events in Atrial Fibrillation (ARISTOTLE) trial, patients on warfarin and amiodarone had lower times within the therapeutic range than patients not on amiodarone (56.5 % versus 63.0 %; p<0.0001) and a significantly increased risk of stroke and systemic embolism.16 In the Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation –Thrombolysis in Myocardial Infarction 48 (ENGAGE AF-TIMI 48) trial, patients randomised to 30 mg (or dose-adjusted to 15 mg) of edoxaban treated with amiodarone at the time of randomisation demonstrated a significant reduction in ischaemic events versus warfarin when compared with those not on amiodarone, while preserving a favourable bleeding profile.17 In contrast, amiodarone had no effect on the relative efficacy and safety of high-dose edoxaban.17
Intake of foods – particularly vegetables containing vitamin K, such as spinach, kale and avocado – and herbal supplements can offset the effect of the daily dose of VKA.18,19 Components of grapefruit and grapefruit juice, such as furanocoumarins, inhibit CYP3A4 activity and can therefore increase plasma levels of VKAs.20,21
Direct Oral Anticoagulants
The four currently-available DOACs are dabigatran, rivaroxaban, apixaban and edoxaban. DOACs are used in a number of clinical settings, including the prevention and treatment of venous thromboembolism and stroke prophylaxis in non-valvular AF. In this review we focus on the latter indication. In clinical studies, these drugs show similar efficacy and safety to warfarin, but are more convenient and do not require meticulous dose adjustment and monitoring to achieve optimal treatment.22–26 To date, no interactions with genetic factors have been reported. However, it is important for physicians to be mindful of any interactions that may alter plasma concentrations of DOACs (Table 1).
Effect of Drugs on the Pharmacokinetics of DOACs
Drugs that induce cell efflux transporter P-glycoprotein (P-gp) and/or CYP450 may decrease DOAC plasma concentrations and increase the risk for thromboembolic events, while drugs that inhibit P-gp and/or CYP3A4 may increase DOAC concentrations and therefore increase bleeding risk.
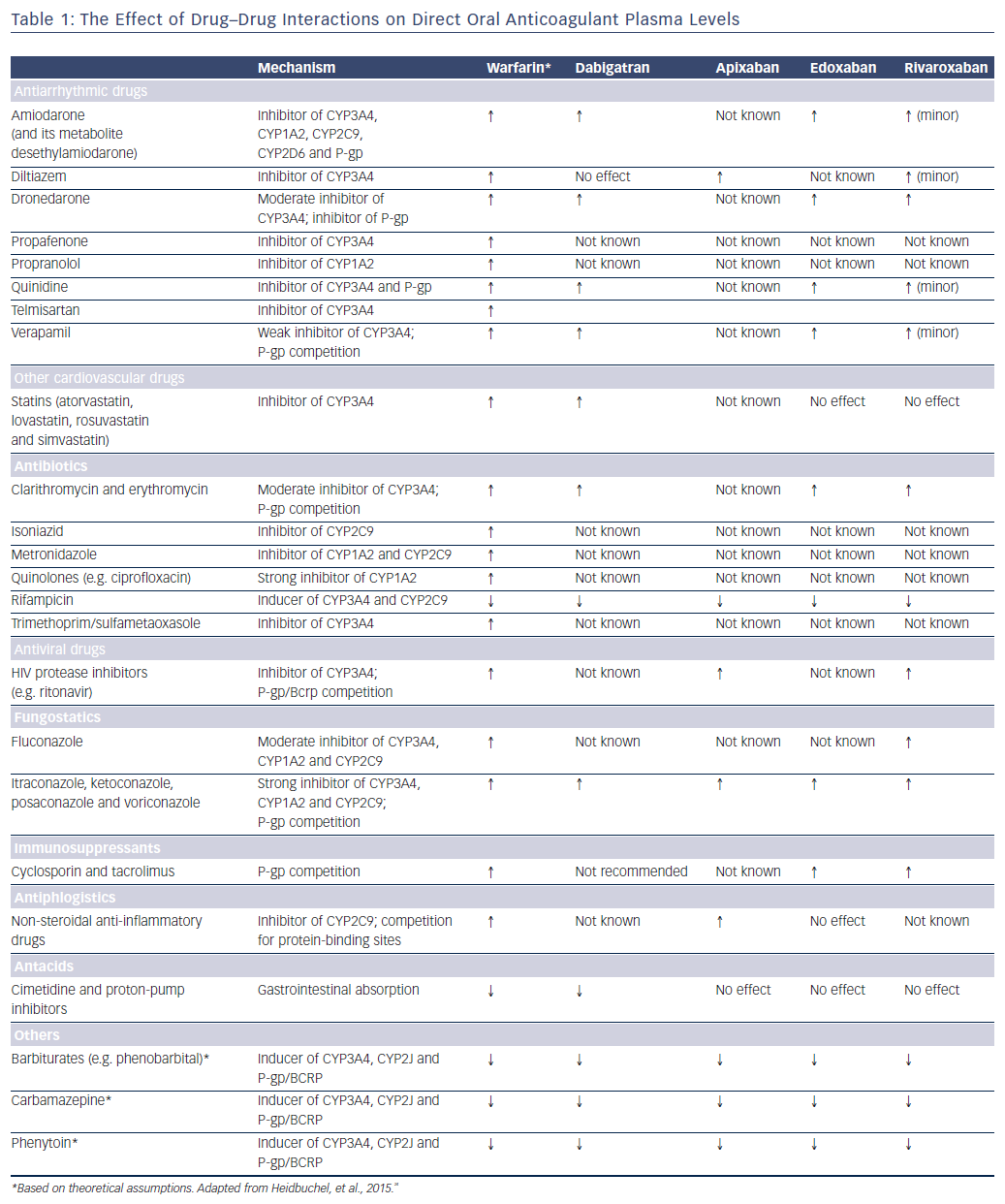
Since dabigatran etexilate is not metabolised by CYP P450 enzymes, it has a low potential for clinically-relevant interactions with drugs metabolised by CYP P450, see Figure 2.25,27 By contrast, this drug is a substrate for P-gp transporters.28 P-gp transporters are efflux transporters that are primarily expressed in the apical/luminal membrane of epithelia of the small intestine, hepatocytes, renal proximal tubules and other sites. P-gp has low substrate specificity and high transport capacity.29 In vitro studies found DDIs between dabigatran and P-gp inhibitors, including amiodarone, clarithromycin, cyclosporin A, itraconazole, ketoconazole, nelfinavir, quinidine, ritonavir and tacrolimus, but no interaction with digoxin.30–32 Co-administration with strong P-gp inhibitors, e.g. ketoconazole, should be avoided.33–35 No dose adjustment is needed with the use of amiodarone, whereas the standard dose of 150 mg twice daily should be reduced to 110 mg twice daily in patients receiving verapamil.36 It has been suggested that the interaction can be minimised if dabigatran is administered 2 hours prior to co-administering any P-gp inhibitor.33
Dabigatran absorption is reduced by the co-administration of anti-acid drugs such as proton-pump inhibitors, although this effect is rarely of clinical relevance.37 Dabigatran bioavailability increases with the concomitant use of ketoconazole or quinidine and decreases with rifampicin,22,38 hence their co-administration should be avoided.
Apixaban and rivaroxaban are all substrates for CYP450, such as CYP3A4, and for P-gp breast cancer resistance protein (Bcrp (ABCG2)) transporters, see Figure 2.36,39 Cytochrome P450 isoenzyme CYP3A4 is a major source of variability in drug pharmacokinetics and response. There are 57 functional human CYPs, but around 10 enzymes belonging to the CYP1, 2 and 3 families are responsible for the biotransformation of most foreign substances, including 70–80 % of all drugs in clinical use; 248 drug metabolism pathways involve CYP.40 Cytochrome P450 (CYP3A4) is involved in the hepatic clearance of rivaroxaban and apixaban to different extents (33 % and 25 %, respectively).39,41 Dabigatran is not a CYP3A4 substrate, and less than 4 % of edoxaban is metabolised via CYP3A4.
Apixaban and rivaroxaban plasma concentrations have been shown to increase to a clinically relevant degree in the presence of ketoconazole and ritonavir (a strong dual inhibitor of CYP3A4 and P-glycoprotein [P-gp]), while erythromycin (a moderate inhibitor CYP3A4 and weak inhibitor of P-gp), clarithromycin (a strong inhibitor CYP3A4 and weak-to-moderate inhibitor P-gp) and fluconazole (a moderate inhibitor CYP3A4, and potentially Bcrp) result in a moderate but not clinically-relevant increase in exposure.32,39,42–44 Co-administration of diltiazem leads to small increases in mean apixaban area under the curve (AUC) and Cmax45,but not rivaroxaban.
Co-administration of ketoconazole or ritonavir leads to 2.6- or 2.5-fold increases in mean rivaroxaban AUC, respectively, and 1.7- or 1.6-fold increases in rivaroxaban Cmax, respectively, and is associated with increased bleeding risk.39,46 Ketoconazole 400 mg leads to approximately 70 % mean inhibition of non-renal (metabolic) clearance of rivaroxaban and 44 % mean inhibition of active renal secretion, whereas ritonavir leads to a reduction in metabolic clearance of approximately 50 % and reduction in active renal secretion of >80 %.39 No significant interactions have been reported following co-administration of rivaroxaban and the CYP3A4 substrates midazolam and atorvastatin.39,47,48 Co-administration of apixaban and rivaroxaban with strong CYP3A4 or P-gp inhibitors, such as ketoconazole or ritonavir, should be avoided.40 There is no need for dose adjustment when co-administered with weak CYP3A4 and or P-gp.
Edoxaban elimination is only slightly dependent on CYP3A4 mechanisms.49,50 Edoxaban exposure is affected by P-gp inhibitors and inducers. Co-administration of amiodarone, quinidine and ketoconazole has been reported to increase exposure to edoxaban.50–52 Erythromycin and cyclosporin also increase edoxaban exposure.52 The amount of edoxaban should be halved when co-administered with P-gp inhibitors that increase edoxaban exposure by ≥1.5 fold (e.g. dronedarone increases exposure by 84.5 %, quinidine by 76.7 % and verapamil by 52.7 %).50 No dose adjustment is needed with amiodarone as it only increases edoxaban exposure by 40 %.53
Digoxin is widely used for ventricular rate control in patients with AF. It is a P-gp substrate but, despite this, no interactions have been reported following co-administration with dabigatran, rivaroxaban and edoxaban.47,48,54 However, in the Rivaroxaban – Once Daily, Oral, Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation (ROCKET-AF) study, digoxin treatment in patients with AF taking rivaroxaban or warfarin was associated with a significant increase in all-cause mortality, vascular death and sudden death.55 Similar data were reported for edoxaban in the ENGAGE-AF TIMI 48 trial.56
Concomitant use of strong CYP3A4 inducers, such as rifampicin, phenytoin, carbamazepine or phenobarbital, can significantly lower DOAC plasma concentrations and significantly reduce the AUC for rivaroxaban, which is thought to cause a parallel decrease in pharmacodynamic effect. Co-administration of rifampicin leads to a decrease of approximately 50 % in the mean AUC of rivaroxaban.36,57,58 Rifampicin has been reported to increase the apparent oral clearance of edoxaban by 33 % and decrease its half-life by 50 %, primarily due to its effect on P-gp, since edoxaban is minimally dependent on CYP3A4.59 Administration of the P-gp inducer rifampicin (which is also a CYP3A4 inducer) for 7 days resulted in a significant reduction in the bioavailability of dabigatran, which returned almost to baseline after 7 days’ washout.59 A reduction in the bioavailability of apixaban has also been reported.60 Anti-epileptic drugs such as carbamazepine, levetiracetam, phenobarbital, phenytoin and valproic acid might also decrease the effect of DOACs by inducing P-gp, but further studies are required to confirm this.31
Effect of Drugs on the Pharmacodynamics of DOACs
Particular caution is needed in patients in whom DOACs are co-administered with antiplatelet agents (e.g. aspirin, P2Y12 inhibitors) and non-steroidal inflammatory drugs, owning to these agents’ influence on haemostasis and increased bleeding risk.60 The co-administration of DOACs should be avoided and/or limited in time, unless specifically recommended.61 Clinical data demonstrating increased bleeding risk when individual DOACs are co-administered with antiplatelet agents are presented below.
In a pooled analysis from the four large randomised controlled trials of DOACs that included 42,411 patients – 33.4 % of which, i.e. 14,148 patients, were also on aspirin or another antiplatelet drug, there was no additional benefitin those taking anticoagulation and antiplatelet therapy for stroke prevention when compared with anticoagulation alone.62 There was, however, an increased risk of bleeding.63,64 Co-administration of aspirin and dabigatran, and of aspirin and apixaban, showed an increased rate of bleeding events. Other studies found that co-administration of a single antiplatelet therapy and edoxaban resulted in higher bleeding rates than in those not receiving single antiplatelet therapy, while co-administration of aspirin and edoxaban showed a two-fold increase in bleeding time.64–68 A similar impact on bleeding events can be expected for rivaroxaban, however there are as yet no published data from the ROCKET-AF trials. Co-administration of rivaroxaban and clopidogrel increased the bleeding time in healthy subjects, but did not affect the pharmacokinetic or pharmacodynamic parameters of either drug.66 Following acute coronary syndrome, apixaban combined with standard antiplatelet therapy (21 % dual antiplatelet therapy) showed a dose-related increase in bleeding events without a significant reduction in recurrent ischaemic events.69,70 A dose-related increase in bleeding events was also reported with rivaroxaban in combination with standard antiplatelet therapy (92 % on dual antiplatelet therapy) in the Anti-Xa Therapy to Lower Cardiovascular Events in Addition to Standard Therapy in Subjects with Acute Coronary Syndrome – Thrombolysis in Myocardial Infarction 52 (ATLAS ACS2-TIMI 52) study.71

The Study Exploring Two Treatment Strategies of Rivaroxaban and a Dose-Adjusted Oral Vitamin K Antagonist Treatment Strategy in Subjects with Atrial Fibrillation who Undergo Percutaneous Coronary Intervention (PIONEER-AF PCI) trial was the first to study the use of a DOAC as an alternative to VKA for patients with non-valvular AF requiring stent implantation.68 Rivaroxaban 15 mg once daily (plus P2Y12 inhibitor) and rivaroxaban 2.5 mg twice daily (plus P2Y12 inhibitor and aspirin 75 mg or 100 mg once daily) were associated with lower risks of clinically-significant bleeding than standard triple therapy (16.8 % and 18.0 % [HR 0.59, 95 % confidence interval [CI] 0.47–0.76, p<0.001], versus 26.7 % [HR 0.63, 95 % CI 0.50–0.80, p<0.001]). However, the trial lacked formal testing of the non-inferiority of the rivaroxaban regimens compared with the warfarin-based triple therapy. The Evaluation of Dual Therapy with Dabigatran versus Triple Therapy with Warfarin in Patients with AF that Undergo a PCI with Stenting (RE-DUAL) clinical study (n=2,725) showed that, among patients with AF who had undergone PCI, the risk of bleeding was lower in those who received dual therapy with dabigatran and a P2Y12 inhibitor than in those who received triple therapy with warfarin, a P2Y12 inhibitor and aspirin (HR 0.52; 95 % CI 0.42–0.63, p<0.001 for non-inferiority, p<0.001 for superiority). Dual therapy was non-inferior to triple therapy in terms of risk of thromboembolic events.72 The use of DOACs in patients with AF that undergo a PCI with stenting is being tested further in two other randomised trials: the Trial to Evaluate Safety of Eliquis (Apixaban) in Nonvalvular Atrial Fibrillation Patients with a Recent Acute Coronary Syndrome or Undergoing Percutaneous Coronary Intervention (AUGUSTUS) and the Edoxaban Treatment versus Vitamin K Antagonist in Patients with Atrial Fibrillation Undergoing Percutaneous Coronary Intervention (ENTRUST-AF PCI).
In the absence of safety data from randomised clinical trials (only 6 % of patients were treated at baseline with ticagrelor or prasugrel in the PIONEER AF-PCI trial) and worrisome bleeding signals in registries, the use of these drugs should be avoided as part of triple therapy (and even double therapy) at this stage.68,69 Patients may need to switch from DOACs to VKA and vice versa. Transition from warfarin to rivaroxaban was found to enhance the prolongation of prothrombin time/INR activity due to a supra-additive effect during the initial transition period; however, there was no effect on anti-factor Xa activity.73 Dabigatran, rivaroxaban, apixaban and edoxaban can be administered safely after enoxaparin.74–76 However, because of their additive effect, co-administration of DOACs with other anticoagulants (e.g. low-molecular-weight heparin) is discouraged.
Labelling recommendations for the concomitant use of DOACs with other drugs are given in Table 2.
Effect of Food on the Pharmacokinetics or Pharmacodynamics of DOACs
Although, in theory, food or herbal inhibitors/inducers of CYP3A4 or P-gp might interfere with the pharmacokinetics of DOACs, no direct evidence of such interactions exist.
St John’s wort, a potent inducer of P-gp and CYP3A4, is expected to lower plasma concentrations of dabigatran (a substrate of P-gp), rivaroxaban and apixaban (substrates of P-gp and CYP3A4). Co-administration should be made with caution with dabigatran and avoided with apixaban and rivaroxaban. Rivaroxaban shows increased bioavailability when taken with food, but there is no interaction for the other DOACs;73,77 therefore rivaroxaban should be taken with food, but this is not necessary for the other DOACs. It has been suggested that grapefruit affects the bioavailability of rivaroxaban but this has not been confirmed in clinical studies.78 No information is available regarding the potential pharmacodynamic interactions of DOACs with foods or herbal medicines.
High-risk Patient Groups
Since each of the DOACs undergoes renal elimination to some extent (dabigatran 80 %, rivaroxaban 33 %, apixaban 25 % and edoxaban 50 %), patients with renal impairment or >75 years may be at a higher risk of bleeding complications, especially if they also have potential DDIs.
Patients with cancer and AF may be at increased risk of thromboembolic events, and also for bleeding complications. The net clinical benefit of DOACs in this patient population is unstudied. DOAC treatment in cancer patients should therefore take into account frailty, platelet count and anaemia, as well as anticipated therapy-induced changes in organ function (especially liver and renal function). Some classes of chemotherapy appear to universally interact with CYP3A4, P-pg or both. These include the antimitotic microtubule inhibitors (e.g. vinca alkaloids and taxanes), tyrosine kinase inhibitors (but not erlotinib, gefitinib and sorafenib), and immune-modulating agents, including glucocorticoids and mammalian target of rapamycin inhibitors (but not everolimus). Conversely, none of the frequently-used antimetabolites, platinum-based agents, intercalating agents or monoclonal antibodies has significant inhibitory or inducing effects on CYP3A4 or P-pg. No clear class effect is seen among the topoisomerase inhibitors, anthracyclines, alkylating agents or anticancer hormonal agents; there is significant heterogeneity in drug interaction potential within each of these medication classes. Chemotherapeutic agents are often used in combination, and the clinical relevance of these combined weak or moderate interactions remain mostly unknown. Potential disruption in absorption due to short gut or malnutrition, which are common issues in the cancer population, are of concern.
HIV-positive patients often require anticoagulation therapy because they are at increased risk of venous thromboembolism or cardiovascular disease.79,80 However, many anti-retroviral agents used to treat HIV, such as nevirapine, efavirenz, saquinavir and ritonavir, are inhibitors/inducers of CYP enzymes and/or P-gp. An increased likelihood of adverse reactions or decreased efficacy of DOAC therapy is therefore an important consideration in this patient population.24,75
Bariatric surgical procedures are a well-established approach to the treatment of morbid obesity, offering sustainable weight loss and a reduction in the risk of conditions related to obesity. Roux-En-Y gastric bypass is one of the most common bariatric surgical treatments. The consequences of this procedure are a 95 % reduction in gastric capacity as well as a reduction in the functional length of the gastrointestinal tract from bypassing the duodenum and proximal jejunum. These changes potentially augment the effect of P-gp induction on limiting drug absorption. Bariatric surgery has been linked to nutritional deficiencies but has not been extensively studied for its effects on DOAC drug absorption and activity.81
Summary
Numerous pharmacokinetic and pharmacodynamic interactions with drugs and food can influence the efficacy and safety of both VKAs and DOACs. Despite fewer food and drug interactions compared with warfarin, physicians should still consider DDIs when prescribing DOACs. Pharmacokinetic DDIs that may occur in association with DOACs are largely mediated by the P-gp efflux transporter protein alone or in combination with CYP3A4 enzymes. In addition to managing pharmacokinetic-based interactions, clinicians should avoid unnecessary pharmacodynamic interactions between DOACs and antiplatelet agents and non-steroidal anti-inflammatory drugs. Due to the extensive renal elimination of some DOACs (particularly dabigatran), DDIs are more significant in patients with renal impairment. It should be noted that, for many potential interactions with medications often used in AF patients for other comorbidities, no data are available. There is a need for further clinical studies and real-world evidence to provide more information about the potential DDIs of DOACs to further optimise their safety profile.